
4例先天性胆汁酸合成障碍2型患儿的临床特征及遗传学分析
2021-08-17 10:14欧阳文献谭艳芳李双杰
临床肝胆病杂志 2021年8期
姜 涛, 欧阳文献, 谭艳芳, 唐 莲, 张 慧, 李双杰
湖南省儿童医院 肝病中心, 长沙 410007
先天性胆汁酸合成障碍2型(congenital bile acid synthesis disorder type 2,CBAS2)是醛酮还原酶家族1D1(Aldo-ketoreductase family1 member D1,AKR1D1)基因突变引起△4-3-氧固醇-5β-还原酶缺乏所致的胆汁酸合成障碍性疾病,其主要临床表现为新生儿胆汁淤积症和暴发型肝衰竭[1]。自1988年Setchell等[2]根据血尿FAB-MS分析首次诊断2例△4-3-氧固醇-5β-还原酶缺陷患儿后,有关CBAS2的病例报道仅数十例[3-4]。现对收治的CBAS2患儿临床特征、转归以及AKR1D1基因变异进行了分析,报道如下。
1 资料与方法
1.1 研究对象 收集2017年1月—2020年11月本院肝病中心诊治的4例CBAS2患儿病例资料,其中男2例,女2例。
1.2 基因检测及分析 在父母签署知情同意书后,抽取患儿和父母外周静脉血2 ml,提取合格的人类基因组DNA进行文库制备。文库制备是将基因组DNA打断成主带<500 bp,峰值在350 bp的小片段DNA,将打断后的DNA片段进行末端补平,在3′端加“A”碱基,使得DNA片段能与5′端带有“T”碱基的特殊接头连接,经Pre-Capture LM-PCR对带有接头的文库进行扩增。等量混合文库后,加入人类外显子文库进行杂交,捕获Exon capture区域,再通过PCR扩增富集捕获后产物。琼脂糖凝胶电泳测定文库的片段大小,Qubit3.0及荧光定量PCR测定文库的浓度。将质检合格的文库稀释至上机浓度,使用IlluminaHiseq测序平台进行测序分析,测序结果通过生物信息学方法进行数据分析,后由遗传咨询师进行报告解读。
检索内部数据库、dbSNP、ESP6500、ExAC等人群数据库,标注单核苷酸多态性和低频良性变异。对患儿父母及主要亲属相应突变外显子进行PCR测序,以明确突变来源。应用预测软件对变异的保守性/致病性/危害性进行预测。检索HGMD、PubMed、Clinvar、OMIM等数据库中的变异相关文献,分析文献内容,参考美国医学遗传学和基因组学学会变异分类指南,对变异进行分类。
1.3 伦理学审查 本研究方案经由湖南省儿童医院伦理委员会审批,批号:KY2020-06。
2 结果
2.1 临床特点和预后 4例CBAS2患儿均以皮肤黄染为首发表现,确诊年龄为3~5月龄,病例2父母为近亲结婚。所有患儿均有肝大、浅黄色大便表现,病例1伴有生长发育迟缓和脾大,实验室检查提示:胆汁淤积,转氨酶升高,凝血全套、Alb和TBA正常(表1),2例患儿伴有脂溶性维生素(A、D、E)缺乏,腹部彩超提示肝大、小胆囊和肝实质弥漫性病变。
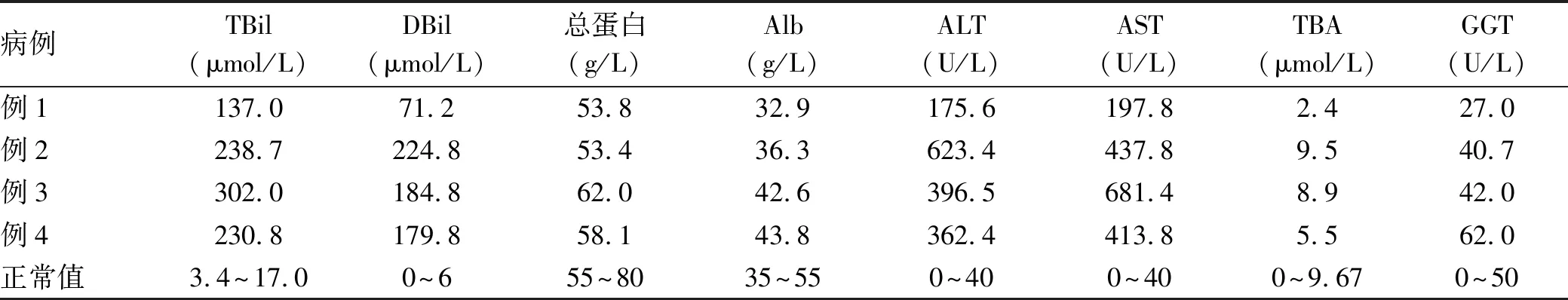
表1 4例CBAS2患儿入院后初次肝功能指标检查结果
患儿入院后予以护肝、熊去氧胆酸(10~15 mg·kg-1·d-1)利胆等治疗后,胆红素仍反复升高,甚至进行性加重。病例3于8月龄时因肝硬化失代偿而死亡。病例1、2和4予以熊去氧胆酸治疗数月后,改为鹅去氧胆酸(5~10 mg·kg-1·d-1)治疗,电话随访病例1(3岁8月龄)和病例4(2岁)患儿肝功能一直正常(黄疸消退时间不详),病例2(6月龄)患儿在使用鹅去氧胆酸治疗1个月后胆红素恢复正常,转氨酶仍有轻度升高。3例患儿的生长发育均达标,但其远期预后需定期随访。
2.2 家系基因分析结果 基因分析结果显示,3例复合杂合突变,1例纯合突变,共5种变异(表2),其中c.797G>A和c.580-13T>A已在多个病例中报道[3,5],而c.70G>C、c.93+2T>C和c.579+2_579+4delinsA这3种新变异在PubMed、OMIM、HGMD、Clinvar等数据库及文献中未见报道,且在正常人数据库(dbSNP、ESP6500和ExAC等)中未见收录,表明不是人群多态性位点,结合临床表现,软件预测提示为致病或可能致病。
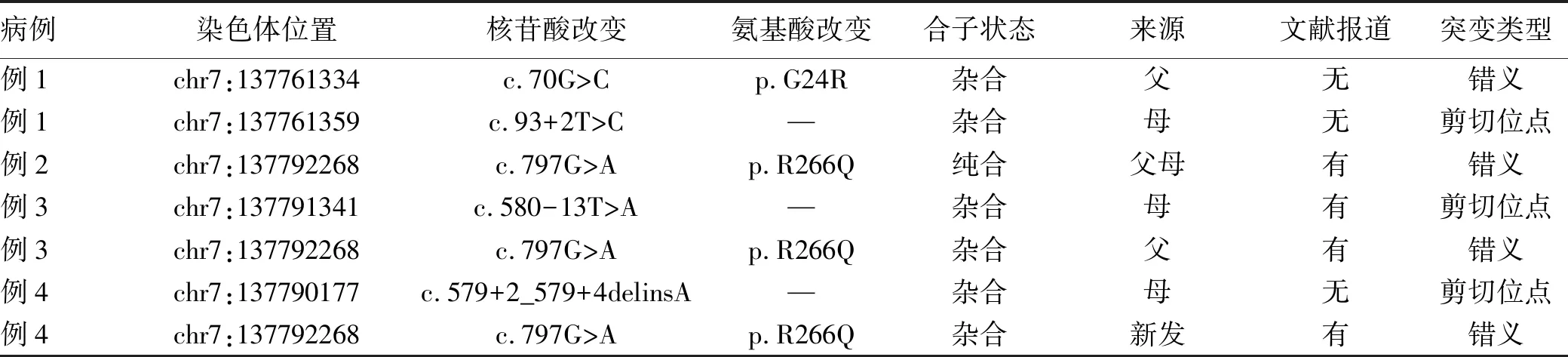
表2 4例CBAS2患儿基因变异情况
3 讨论
胆固醇在肝细胞内经过一系列的酶促反应后形成胆汁酸,其合成过程中至少有16种酶参与,任何一种酶的缺乏均可导致正常胆汁酸生成障碍,从而引起先天性胆汁酸合成障碍,该类疾病均为常染色体隐性遗传病。患儿因肝细胞内不正常的毒性胆汁酸堆积、正常胆汁酸生成减少等而致胆汁淤积、肝衰竭和肝硬化[1,6],同时可出现脂肪和脂溶性维生素吸收障碍等,占儿童胆汁淤积性肝病的1%~2%[7],最常见的为CBAS1和CBAS2[7],早期初级胆汁酸的治疗可明显改善患儿临床症状,减少肝移植[8]。
CBAS2是由于胆汁酸合成过程中的△4-3-氧固醇-5β-还原酶缺乏,导致其底物转化为7α-羟3-氧-4-胆烷酸、7α、12α-双羟-3-氧-4-胆烷酸、异鹅去氧胆酸和异胆酸,经结合形成含3-氧-△4结构的不典型胆汁酸,进入血液后通过肾脏排出体外[2]。CBAS2患儿最常见的临床表现为新生儿胆汁淤积,其他表现为:黑尿、白陶土样便、脂肪泻、脂溶性维生素缺乏、肝大、生长发育迟缓等,无明显瘙痒症状[9]。生化检查可见高结合胆红素血症,转氨酶增高,GGT轻度升高或正常,TBA正常,凝血功能异常等。肝脏病理检查可见胆管排列紊乱,肝巨细胞样变及肝细胞内胆汁淤积、纤维化、假小叶形成,伴或不伴髓外造血[10-11]。本研究中患儿以胆汁淤积和肝大为主要表现,部分患儿伴生长发育迟缓、脾大和脂溶性维生素缺乏。生化指标提示胆汁淤积,AST水平升高幅度大于ALT,TBA正常,与文献[3]报道一致。病例2为纯合突变患儿,但其临床症状并不比复合杂合突变患儿重,与Drury等[12]报道不一致。值得注意的是,4例患儿在予以熊去氧胆酸治疗后,TBA和GGT可出现一过性升高,TBA最高可达346.7 μmol/L,GGT可达337 U/L。因此,对于曾使用熊去氧胆酸治疗的患儿,TBA和GGT的一过性升高可能导致临床上的误诊或漏诊,需反复多次监测GGT和TBA的变化。同时需与低GGT型胆汁淤积性肝病如进行性家族性肝内胆汁淤积症[13]、关节畸形-肾小管功能不全-胆汁淤积综合征和良性复发性肝内胆汁淤积症等遗传性疾病鉴别,若患儿GGT和TBA均正常,则需考虑先天性胆汁酸合成障碍。
临床上高度怀疑CBAS2的患儿,可进行尿液胆汁酸谱分析,注意避免在肝衰竭或严重肝损害时进行,需结合AKR1D1基因检测[10-11]明确。AKR1D1基因定位于7号染色体q32~33区,含有9个外显子,全长42 kb,在肝脏中高度表达,其编码含有326个氨基酸的△4-3-氧固醇-5β-还原酶,除了参与胆汁酸合成,还参与调控多种CYP家族酶类表达[12],在类固醇激素的降解代谢中起一定作用。目前报道的基因突变为:c.797G>A、 c.919C>T、 c.158A>G、c.396C>A、c.722A>T、c.622C>T、 c.385C>T、 c.467C>G、 c.850C>T、 c.737G>A、c.217C>T、c.853C>T、c.580-13T>A、c.64G>A、c.580-13T>A、c.579+2T>-、c.919C>T、c.587delG、c.579+2delT、c.579+4delC、c.933delG和c.511delT等。以错义突变为主,此外还有无义、缺失和剪切位点突变[14-15]。本研究中基因检测发现,患儿均有AKR1D1基因突变,3例复合杂合突变,1例纯合突变,共5种突变,c.797G>A和c.580-13T>A为已知致病突变[3,5],60%(c.70G>C、c.93+2T>C和c.579+2_579+4delinsA8)的新突变主要为错义突变(2/5)和剪切位点突变(3/5)。错义突变c.70G>C,蛋白质结构预测可能有害;经典剪切位点突变c.93+2T>C,生物危害性较高;c.579+2_579+4delinsA该变异位于mRNA剪接区域,核苷酸序列高度保守,并且多种计算辅助算法均预测此变化可能会影响蛋白功能。结合患儿的临床表现和家系分析,可诊断为CBAS2。
鹅去氧胆酸为人体必需的初级胆汁酸,可通过负反馈机制抑制CYP7A1表达,下调异常胆汁酸的合成,减少其对肝细胞的毒害作用。大量文献报道,熊去氧胆酸对CBAS2患儿治疗效果欠佳,而鹅去氧胆酸可有效缓解临床症状,避免进展性肝损害。临床上对于口服药物治疗效果欠佳或者肝衰竭、肝硬化者,可考虑肝移植。本研究中4例CBAS2患儿初期予以熊去氧胆酸治疗,效果欠佳,其中1例患儿8月龄时死亡,3例患儿在使用鹅去氧胆酸治疗后胆红素恢复正常,与文献[16]报道一致。对于合并脂溶性维生素缺乏的患儿可予以补充脂溶性维生素,同时应警惕糖皮质激素的应用,因其可下调AKR1D1的表达和活性。此外,AKR1D1的下调改变了多个核激素受体的激活,以驱动葡萄糖生成和糖原合成基因表达谱的变化,这可能加剧外源性糖皮质激素的不良影响[17]。
总之,临床上对于高度怀疑CABS2的患儿应进行基因检测或者尿液质谱分析,早期诊断并尽早应用鹅去氧胆酸可明显改善临床症状,减少肝移植发生率;同时本研究中3种AKR1D1基因新变异的识别丰富了AKR1D1基因谱。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:姜涛负责课题设计,资料分析,撰写论文;谭艳芳、唐莲、张慧参与收集数据,修改论文;李双杰、欧阳文献负责拟定写作思路,指导撰写文章并最后定稿。
猜你喜欢
基层中医药(2022年7期)2022-11-17
现代临床医学(2022年4期)2022-09-29
保健医苑(2022年5期)2022-06-10
新疆医科大学学报(2022年1期)2022-01-27
珠江水运(2021年15期)2021-08-29
中国现代医生(2021年9期)2021-07-07
文萃报·周二版(2021年11期)2021-04-06
家庭医学(2018年2期)2018-07-14
黄河黄土黄种人(2017年11期)2017-11-27
水能经济(2017年6期)2017-10-19
