
三维g-C3N4/NixP复合光催化剂的制备及其可见光下产氢性能的研究
2021-08-16 07:51王世强康自虎郑言贞
北京化工大学学报(自然科学版) 2021年4期
王世强 康自虎 郑言贞 陶 霞
(北京化工大学 有机无机复合材料国家重点实验室, 北京 100029)
引 言
由二维的超薄纳米薄片组装而成的三维结构的g-C3N4(3D g-C3N4)光催化剂,不仅拥有大的比表面积,而且可以在光催化过程中利用入射光子在开放框架内的多重反射来增强光利用率[1]。更为重要的是,三维结构可以作为支撑有效防止超薄纳米片因范德华力引起的团聚现象,为电子运输提供通路[2]。目前,制备3D g-C3N4的方法可分为模板法与非模板法。使用模板法制备g-C3N4的过程中,往往需要复杂的前驱体灌注和利用有害的化学试剂除去模板的步骤,整个过程不仅复杂而且会对环境带来破坏[3]。超分子自组装法作为一种非模板法,可以通过改变自组装过程的条件对氮化碳的构成、形貌和光电化学性质进行优化,进一步促进其光催化性能提升[4-6],而且这种方法还具有过程简单、原料价格低廉、环境友好等优点[7]。
考虑到贵金属作为g-C3N4光催化产氢助催化剂会受到其稀缺性和高成本的制约,因此发展低成本的光催化产氢助催化剂是有必要的[8]。Liu等[9]通过密度泛函理论预测了过渡金属磷化物具有作为高活性产氢助催化剂的潜力。此后,过渡金属磷化物凭借其低过电位和高稳定性,开始作为有效的助催化剂应用于g-C3N4光催化产氢中[10-12],这些都证明了过渡金属磷化物具有广阔的应用前景。
对于制备过渡金属磷化镍负载的3D g-C3N4,选用合适的负载方式是关键。通过表面磷化的方法往往需要繁琐的步骤,并释放出剧毒的磷化氢(PH3)气体,甚至会使氮化碳结构内出现不可定量的磷元素的掺杂,带来不利的影响[13]。采用物理混合的自组装法得到磷化镍修饰的3D g-C3N4,可能会导致其三维结构的表面松弛或坍塌,引起电子传递受阻,并且这种方法不易使助催化剂均匀分散在3D g-C3N4内[14]。相比自组装法,原位负载法已经被证明是一种更优的负载方式[15]。目前,在维持3D g-C3N4形貌的基础上采用原位光沉积的方式负载非贵金属助催化剂并应用于光催化产氢领域的研究较少,因此本文通过氢键超分子自组装策略制备了3D g-C3N4,并采用原位光沉积法在3D g-C3N4上负载过渡金属磷化物助催化剂NixP,制备出3D g-C3N4/NixP复合光催化产氢催化剂,并采用多种手段对其进行了表征;以三乙醇胺为牺牲剂,测试了3D g-C3N4/NixP复合光催化剂的光催化产氢性能和循环稳定性,为光催化产氢催化剂的设计与制备提供了一个新思路。
1 实验部分
1.1 实验原料
三聚氰酸,纯度98%,麦克林公司;三聚氰胺、三乙醇胺、六水合硫酸镍(NiSO4·6H2O)、次磷酸钠(NaH2PO2),纯度99%,阿拉丁试剂有限公司;去离子水,实验室自制。
1.2 复合催化剂的制备
1.2.13D g-C3N4
通过超分子自组装策略制备3D g-C3N4。称取1.26 g三聚氰胺和1.29 g三聚氰酸,分散在50 mL去离子水中,磁力搅拌12 h,将悬浊液离心,得到白色沉淀物,冷冻干燥96 h。将得到的超分子前驱体放入带盖的瓷坩埚中,用铝箔包裹后形成半密封体系。将半密封的坩埚放入马弗炉中,以5 ℃/min的升温速率升温至600 ℃,并保持4 h,使体系自然冷却即得到3D g-C3N4,标记为DCN,制备过程如图1(a)所示。
作为对比,通过传统的热聚合法制备了体相g-C3N4。称取1.26 g三聚氰胺代替超分子前驱体,按照相同的方法将坩埚体系放入马弗炉中,采用体相g-C3N4常规的合成条件,以5 ℃/min的升温速率升温至550 ℃,保温4 h,待体系自然降温得到体相g-C3N4,标记为BCN。
1.2.23D g-C3N4/NixP
采用原位光沉积法制备3D g-C3N4/NixP。称取30 mg 3D g-C3N4,在产氢试管中与2 mL去离子水混合,然后与4.0 mL 0.1 mol/L NiSO4水溶液和4.0 mL 0.7 mol/L NaH2PO2水溶液混合。在持续的搅拌下用氮气吹扫混合系统40 min,随后将混合系统在300 W氙灯(紫外- 可见光)下照射。照射结束后,使用离心机在低速下分离反应液和产物,然后用去离子水多次冲洗,在2 000 r/min下离心5 min,收集产物,冷冻干燥12 h,获得3D g-C3N4/NixP复合光催化剂,制备过程如图1(b)所示。其中NixP的沉积量可以通过改变光照时间进行调整,光照10、20、30、40 min得到的复合光催化剂分别命名为DCN- 10、DCN- 20、DCN- 30、DCN- 40。使用相同的光沉积方法制备了BCN与NixP复合的BCN/NixP光催化剂,光照20 min得到的光催化剂命名为BCN- 20。

图1 多孔3D g-C3N4纳米片与多孔3D g-C3N4/NixP复合光催化剂的制备流程Fig.1 Preparation process of porous 3D g-C3N4 nanosheet and porous 3D g-C3N4/NixP composite photocatalyst
1.3 表征与测试
使用扫描电子显微镜(SEM)(JSM- 6701F型,日本电子株式会社)、透射电子显微镜(TEM)(Hitachi 7700型,日本日立科学仪器有限公司)以及高分辨透射电子显微镜(HRTEM)(Hitachi 9500型,日本日立科学仪器有限公司)表征催化剂的微观形貌和纳米结构;使用X射线衍射仪(XRD)(Rigaku D/max- 2500型,日本理学株式会社)表征催化剂的晶体构型,采用Cu Kα辐射(λ=0.154 06 nm),扫描范围5°~70°,扫描速率5(°)/min;使用傅里叶变换红外光谱仪(FT- IR)(Bruker Vertex 70V型,德国Bruker公司)分析催化剂中的官能团与化学键的振动情况,测试范围500~4 000 cm-1;使用X射线光电子能谱仪(XPS)(ESCALAB 250型,美国ThermoFisher Scientific公司)测试催化剂表面各元素组成与成键情况,采用Al Kα辐射,以碳单质的标准峰位284.8 eV进行校正;使用紫外可见分光光度计(Lambda 950型,美国Perkin Elmer公司)测试催化剂的光吸收特性,以BaSO4作为基底,扫描范围250~800 nm。
1.4 光催化产氢性能及稳定性测试
称取5 mg光催化剂,加入到装有10 mL 20%(体积分数)三乙醇胺水溶液(牺牲剂)的石英管反应器中,然后在搅拌下向试管中通入高纯度N2,保持20 min以尽可能排除试管中的空气。然后以配备有滤波片(λ≥420 nm)的300 W氙灯(CEJ- HXF300型,北京中教金源科技有限公司)作为光源进行光照,每隔30 min取样,产生的H2通过以N2作为载气的气相色谱仪(7890B型,安捷伦公司)进行检测,每个样品平行重复3次,结果以平均值±标准差表示。在光催化循环试验中,每进行一次循环试验后向反应器中补充一定量的三乙醇胺。产氢速率以单位时间内单位质量催化剂产生的氢气的物质的量表示,μmol/(g·h);产氢量以单位质量催化剂产生的氢气的物质的量表示,μmol/g。
2 结果与讨论
2.1 表征结果
2.1.1微观形貌
图2为BCN、DCN与DCN- 20的微观形貌表征图。由图2(a)可以看出BCN呈现出堆叠的块状形貌;由图2(b)可以看出DCN呈现出由多孔纳米片组装而成的三维开放框架形貌;图2(c)表明DCN- 20的形貌同样保持了三维开放框架的形貌特征。通过图2(d)进一步发现,BCN由于堆叠的块状结构,其TEM图像显示出一片阴影;由图2(e)与2(f)可以观察到DCN与DCN- 20具有由卷曲的超薄片构成的三维框架形貌。由图2(f)还可以进一步看出,在构成三维形貌的超薄纳米片上均匀分布着直径约几十纳米的NixP颗粒;通过图2(g)的HRTEM图进一步观察,未发现明显的单晶晶格条纹,在图2(g)的选区电子衍射图(SAED)中可以看出NixP的电子衍射条纹为弥散的多晶衍射环,说明通过光沉积法制备的NixP为无定形。

图2 BCN、DCN与DCN- 20样品的SEM与TEM图Fig.2 SEM and TEM images of BCN, DCN and DCN- 20
2.1.2晶体构型
图3为BCN、DCN与DCN- 20的XRD谱图。由图3可以看出,3个样品都表现出标准的石墨相氮化碳的峰,其中位于13°左右较弱的衍射峰和位于27°左右较强的衍射峰分别对应g-C3N4的(100)和(002)晶面,这表明NixP的引入不会改变DCN的基本结构。进一步观察可以发现DCN与DCN- 20样品的(002)晶面的特征峰出现的位置较BCN稍微向右偏移,表明所制备的3D g-C3N4的界面堆垛距离减小,而(002)晶面的峰强度较BCN明显减弱则是由于平面尺寸减小造成的。另外在测试中没有出现NixP的峰,进一步证明所制备的NixP为非晶态,这与HRTEM图中未发现晶格和SAED图中呈现弥散的多晶衍射环结果一致。

图3 BCN、DCN与DCN- 20的XRD谱图Fig.3 XRD patterns of BCN, DCN and DCN- 20
2.1.3FT-IR结果
由图4的红外吸收光谱可以发现,3个样品的吸收峰位置基本一致,表明3个样品都具有g-C3N4的碳氮杂环结构。其中,810 cm-1附近的尖峰对应于庚嗪环结构的伸缩振动;1 200~1 600 cm-1附近的吸收峰归因于芳香族C—N杂环的伸缩振动;在3 000~3 300 cm-1之间的宽峰对应于反应过程中未缩合的—NH2或吸附于样品表面的H2O产生的O—H伸缩振动;DCN和BCN的出峰位置一致,表明三聚氰酸在聚合过程中完全参与反应,基本不会对g-C3N4平面内原有的碳氮杂环结构造成破坏。样品DCN- 20的峰与前两者一致则证明NixP的引入不会破坏DCN具有的g-C3N4分子结构。

图4 BCN、DCN与DCN- 20的FT- IR谱图Fig.4 FT- IR spectra of BCN, DCN and DCN- 20
2.1.4元素分析

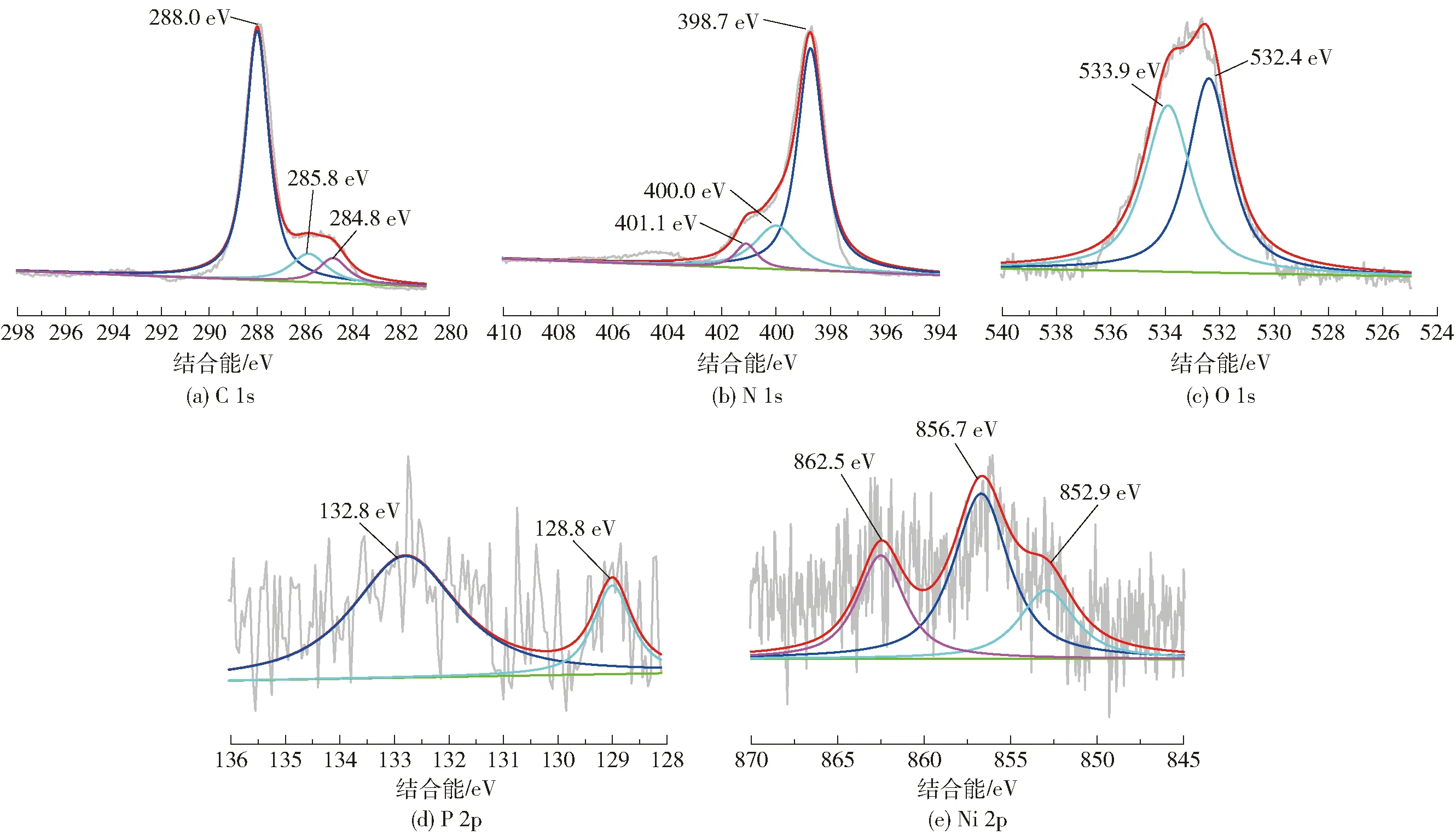
图5 DCN- 20样品的高分辨XPS谱图Fig.5 High resolution XPS spectra of DCN- 20
2.1.5光吸收特性
图6(a)为不同催化剂的紫外可见漫反射光谱(UV- Vis DRS)谱图,从图中可以看出BCN表现出约450 nm的本征吸收边,与文献报道的结果[16]相近。与BCN相比,DCN与DCN- 20的本征吸收边发生了蓝移现象,原因是构成DCN与DCN- 20的g-C3N4纳米片单元的薄层结构引起的量子限域效应,这在关于纳米片结构的g-C3N4的报道中是常见现象[17-18]。对比样品的吸光能力可以发现,DCN在可见光区的光吸收能力大于BCN,负载NixP后的DCN- 20样品的吸光度进一步增大。根据图6的带隙能谱图(Kubelka- Munk图)计算出BCN的带隙能为2.73 eV,与文献报道的值[16]相近。由于蓝移现象,DCN的带隙能较BCN增大到2.97 eV。利用原位光沉积法制备出的DCN- 20光催化剂的带隙能为2.82 eV,吸收边较DCN发生红移,且吸光度明显增强,这是由于NixP的负载提高了复合材料的光吸收能力所致。

图6 BCN、DCN与DCN- 20的UV- Vis DRS与带隙能谱图Fig.6 UV- Vis DRS and corresponding band gap energy spectra of BCN, DCN and DCN- 20
2.2 光催化产氢性能
图7为不同光催化剂的产氢速率。从图中可以看出,BCN的产氢速率为痕量,未负载助催化剂的DCN的产氢速率为7 μmol/(g·h),它们在光催化产氢测试中都显示出极低的产氢速率。对于光照不同时间得到的3D g-C3N4/NixP复合光催化剂,可以看出光照20 min得到的DCN- 20产氢速率最大,可达1 720 μmol/(g·h)。在光沉积的过程中,光照时间过短会导致NixP助催化剂的负载量不足;当光照时间增加时,随着NixP助催化剂负载量的增多,光催化产氢速率会有所增加;当光照时间进一步增加时,过长的光照时间会带来过大的NixP助催化剂负载量,这样会产生掩蔽效应而使得复合光催化剂的产氢性能下降。作为对比,我们通过同样的光沉积方法制备了BCN与NixP的复合样品BCN- 20,并将其光催化产氢速率与DCN- 20进行对比。可以发现,BCN由于堆叠的块状结构,其暴露的有效活性面积较小,因此BCN- 20并没有表现出优异的光催化产氢性能,其产氢速率仅为15 μmol/(g·h),远远小于DCN- 20样品的光催化产氢速率。

图7 不同光催化剂的产氢速率Fig.7 Hydrogen production rates of different catalysts
2.3 光催化稳定性
为了测试DCN- 20样品的光催化产氢稳定性,对其进行了光催化循环产氢试验,结果如图8所示。通过光沉积法制备的DCN- 20在经历5个循环的产氢测试后,其产氢能力仅出现了很小的下降趋势,表明DCN- 20样品具有较好的光催化循环稳定性。复合催化剂循环性能下降的原因可能是由于三乙醇胺和Ni2+的络合作用导致的NixP在DCN表面缓慢脱落。

图8 DCN- 20催化剂的光催化产氢循环测试Fig.8 Photocatalytic hydrogen production cycle tests for DCN- 20 catalyst
3 结论
(1)通过超分子自组装策略制备出三维框架结构的3D g-C3N4,为了避免其他负载助催化剂的方式对其形貌造成破坏,采用原位光沉积法在3D g-C3N4上沉积NixP助催化剂。形貌表征结果发现3D g-C3N4/NixP复合光催化剂呈现三维框架形貌,颗粒直径为几十纳米的NixP均匀负载在3D g-C3N4上,并且NixP呈现出无定形;UV- Vis DRS结果表明,与体相g-C3N4和3D g-C3N4相比,3D g-C3N4/NixP复合光催化剂的可见光吸收能力明显增强。
(2)使用光照20 min得到的3D g-C3N4/NixP复合光催化剂在可见光下进行产氢性能测试,结果表明,与未经NixP修饰的3D g-C3N4和体相g-C3N4/NixP相比,其产氢速率显著提高,达到1 720 μmol/(g·h);在经历5个循环的产氢试验后催化性能基本保持稳定,表明复合光催化剂有着良好的光催化稳定性。
猜你喜欢
化学与粘合(2022年5期)2022-10-31
疯狂英语·新阅版(2021年9期)2021-10-30
科学导报(2020年70期)2020-11-09
科学导报(2019年42期)2019-09-03
中国科技纵横(2019年3期)2019-03-25
当代陕西(2018年9期)2018-08-29
分析化学(2017年12期)2017-12-25
科技资讯(2017年24期)2017-09-15
科学与财富(2016年28期)2016-10-14
哈尔滨理工大学学报(2015年5期)2016-01-19
