
盐酸埃罗替尼的合成工艺改进
2021-08-13 06:16:56赵臣康蔡志强
合成化学 2021年6期
赵臣康, 蔡志强*, 李 帅
(1. 沈阳工业大学 石油化工学院辽宁省芳烃下游精细化工专业技术创新中心,辽宁 辽阳 111003;2. 山东省药学科学院 化学药物重点实验室,山东 济南 250101)
盐酸埃罗替尼[1]是由美国OSI公司与罗氏旗下基因泰克公司共同研制开发的一种抗肿瘤新药[2]。2004年11月18日通过FDA审批在美国上市[3],2005年9月19日获欧盟批准上市,主要作为二线或三线药物用于治疗晚期或其它治疗方案无效的非小细胞性肺癌(NSCLC)患者。2005年9月13日,FDA批准其用于胰腺癌的治疗。2007年美国罗氏公司宣布盐酸埃罗替尼在中国上市。2013年,FDA批准埃罗替尼作为携带EGFR突变转移的非小细胞肺癌的一线治疗药物。埃罗替尼靶向可逆并选择性作用于酪氨酸激酶受体的表皮生长因子受体亚型(EGFR-TK),其作用机制是在细胞内与底物竞争,抑制EGFR-TK磷酸化,阻断肿瘤细胞信号的传导,抑制肿瘤细胞的生长,诱导其调亡[4]。临床研究显示该药对多种肿瘤有抑制作用,与传统的化疗药物相比,具有不良反应较轻,安全性和耐受性较好等优势。
埃罗替尼的合成路线已有较多报道[5-17],归纳后主要包含以下 3条合成路线:(1)在文献[5]报道的合成路线中,酚O-烷基化所采用的丙酮经脱气处理,且反应结束后,加入大量的乙醚使生成的溴化钾完全析出,操作较为繁琐,乙醚的使用不利于放大生产的要求;在硝基还原时,采用了价格昂贵的催化剂PtO2·H2O,且需要加压(3.2 bar),对设备要求较高,成本高;环合产物需精制,且所用混合溶剂用量较大;在氯代反应时,草酰氯毒性大,对操作条件要求高;(2)文献[7]采用6,7-二甲氧基喹唑啉-4-酮为原料,经过7步反应得产品。该路线步骤较长,起始原料价格较贵,不宜大规模生产时采用;(3)文献[9]采用的合成条件是以3,4-二羟基苯甲醛为起始原料,原料较贵、步骤较长,共需8步反应,环合生成喹唑啉环时收率只有44%,不宜采用。
本文在文献[5-21]方法基础上,设计了一条新的合成路线。以2-氨基-4,5-双(2-甲氧基乙氧基)苯甲酸乙酯(1)为起始原料,经环合、氯化,然后胺化,成盐精制得盐酸埃罗替尼(4, Scheme 1)。此方法操作简单,反应条件温和,收率高(总收率为72.9%),纯度高(每步纯度均大于99%),产品易于纯化,适合工业化生产。

Scheme 1
1 实验部分
1.1 仪器与试剂
SGWX-4型显微熔点仪; Bruker ARX-400 MHz型核磁共振仪(MeOD和CDCl3为溶剂,TMS为内标);Perkin-Elmer Spcetrum-FTIR型红外光谱仪(KBr压片);Agient1100型四极杆液质联用仪;LC-15C型液相色谱仪。
所用试剂均为分析纯,国药集团化学试剂有限公司。
1.2 合成
(1) 中间体2的合成
在5 L反应釜中,加入1(1.0 kg, 3.2 mol),甲酸胺(0.2 kg, 3.2 mol)和甲酰胺(0.72 kg, 16.0 mol),于120 ℃反应15 h。反应毕,自然降温,于80 ℃时加水1 kg,冷却至0 ℃,搅拌1 h,过滤,滤饼加入2 L乙酸乙酯,搅拌,过滤,滤饼真空干燥得中间体20.81 kg,收率86.24%,纯度99. 12%, m.p.188.2 ~190.1 ℃;1H NMR(400 MHz, CDCl3)δ: 11.726(s, 1H), 8.028(s, 1H), 7.607(s, 1H), 4.300~4.274(m, 6H), 3.870~3.838(m, 6H), 3.482(d,J=0.88 Hz, 6H);13C NMR(100 MHz, CDCl3)δ: 162.55, 155.25, 149.16, 145.66, 142.56, 116.04, 109.57, 107.01, I70.91, 70.74, 68.90, 68.77, 59.55, 59.52; IRν: 3040.10, 2874.18, 2805.10, 1675.66, 1611.44, 1513.69, 1496.46, 1449.04, 1287.80, 1084.44, 890.22 cm-1; MS(ESI)m/z: 295.1{[M+H]+}。
(2) 中间体3的合成
氮气保护下,在50 L反应釜中依次加入二氯乙烷(30 kg),DMF(0.4 kg)和中间体2(0.57 kg, 1.94 mol),降温至0 ℃,缓慢滴加POCl3(0.36 kg, 2.33 mol),滴毕,反应5 h。滴加5%NaOH溶液(10 kg),滴毕,搅拌2 h;分液,蒸干有机相,加入正庚烷搅拌1 h,过滤,滤饼真空干燥得0.56 kg中间体3,收率92.45%,纯度99.24%; m.p.107.5~109.6 ℃;1H NMR(400 MHz, CDCl3)δ: 8.855(s, 1H), 7.432(s, 1H), 7.328(s, 1H), 4.348~4.330(m, 4H), 4.348~4.322(m, 4H), 3.503(s, 3H), 3.492(s, 3H);13C NMR(100 MHz, CDCl3)δ: 159.07, 156.36, 152.48, 150.95, 149.02, 119.53, 107.76, 104.16, 70.55, 70.34, 68.89, 68.81, 59.38; IRν: 2986.85, 2921.32, 2896.26, 2822.02, 1610.83, 1557.11, 1508.80, 1458.62, 1256.78, 1231.96, 1047.50, 865.86, 707.51 cm-1; MS(ESI)m/z: 313.1{[M+H]+}。
(3)4的合成
在氮气保护下,于50 L反应釜中依次加入中间体3(0.80 kg, 2.56 mol)和20.0 kg无水乙醇,加热到60 ℃,再加入间乙炔苯胺(0.33 kg, 2.8 mol),加热回流,搅拌6 h。反应毕,降至0 ℃搅拌2 h,过滤,用1 kg乙醇洗涤。将固体加入3 kg乙醇,再加入8 mL盐酸溶液(6 N),调节pH≈3,搅拌1 h;冷却至0 ℃搅拌2 h;过滤,滤饼真空干燥得41.0 kg,收率91.4%,纯度99.84%; m.p.221.8~223.4 ℃;1H NMR(400 MHz, MeOD)δ: 8.700(s, 1H), 8.109(s, 1H), 7.870(s, 1H), 7.704~7.712(m, 1H), 7.484~7.445(t,J=7.6, 7.8 Hz, 1H), 7.436~7.413(m, 1H), 7.260(s, 1H), 4.007~4.369(m, 4H), 3.891~3.862(m, 4H), 3.607(s, 1H), 3.475~3.475(d,J=4.0 Hz, 6H);13C NMR(100 MHz, MeOD)δ: 160.22, 158.60, 152.21, 149.74, 138.42, 137.03, 131.31, 130.27, 128.97, 126.06, 124.62, 108.95, 105.35, 101.72, 83.65, 79.64, 71.79, 71.71, 70.73, 70.58, 59.59; IRν: 3271.31, 3074.2 8, 2955.36, 2810.30, 1610.58, 1575.22, 1514.35, 1449.07, 1367.34, 1279.47, 1193.42, 1166.35, 878.29, 796.98, 702.68 cm-1; MS(ESI)m/z: 430.1{[M+H]+}。
2 结果与讨论
2.1 中间体2的合成
在中间体2的合成过程中,文献报道的常见环合剂为:乙酸甲脒[11],甲酸铵[5],甲酰胺[18],甲酰胺与甲酸铵混合物[5-6,16]。本文起初尝试单独使用甲酰胺为环合剂,与原料1反应制备中间体2。结果表明:单独使用甲酰胺虽然可以得到中间体2,但是反应温度过高(150 ℃),收率较低(63.8%),甲酰胺的用量过多会造成部分产品溶解于甲酰胺中,甲酰胺的用量过少不利于反应进行。因此,借鉴文献[16]的方法,采用甲酰胺与甲酸铵混合物为环合剂与原料1反应制备中间体2,但文献[16]的方法存在以下不足:甲酰胺的使用量过多,原料摩尔比为n(1)/n(甲酰胺)/n(甲酸铵)=1/30/1;反应温度较高(165 ℃);后处理时使用大量氯仿进行萃取。本文对反应条件进行优化,结果见表1。合成中间体2的最佳反应条件为:n(1)/n(甲酰胺)/n(甲酸铵)=1/5/1,反应时间为15 h,反应温度为120 ℃,收率达86.24%,纯度为99.12%。
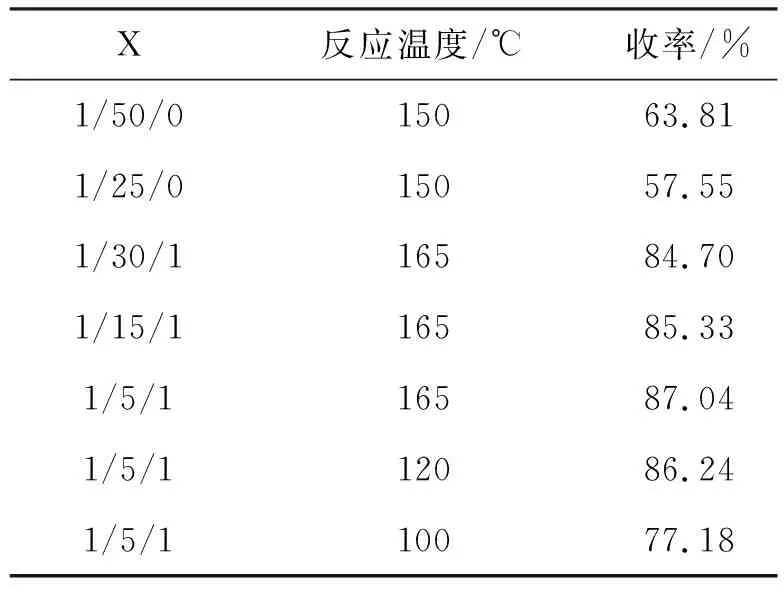
表1 反应条件对中间体2收率的影响*
*X=n(1)/n(甲酰胺)/n(甲酸铵)。
在中间体2的纯化过程中,文献[16]采用有毒溶剂已腈进行重结晶,本文对其进行改进和优化。从异丙醚、乙酸乙酯、甲基叔丁基醚、甲苯4种溶剂中进行筛选,选择出合适的溶剂进行重结晶纯化(表2)。由表2可知,乙酸乙酯作为纯化溶剂较为适宜。

表2 纯化溶剂对中间体2收率和纯度的影响
2.2 中间体3的合成
在中间体3的合成过程中,文献[11,16]均采用氯仿作为反应溶剂,虽然文献[16]较文献[11]氯仿的使用量缩小了一倍,但是氯仿毒性相对较大,不利于工业化大规模生产,故本文采用较低毒性的二氯乙烷作为反应溶剂。文献报道选用草酰氯[5-8]作为氯化试剂,且用量比例较大,草酰氯因具有高毒性和腐蚀性,不适合工业上大规模生产;另据文献报道选用氯化亚砜[18-19]为氯化试剂,在尝试氯化亚砜作为氯化试剂进行氯化[n(中间体2)/n(氯化亚砜)=1/1.2]时,产物收率极低,可能是由于酚羟基活性低,低量的氯化亚砜达不到较好的效果,若加大氯化亚砜的用量,则又不利于环保。最终选用三氯氧磷作为氯化试剂与中间体2反应,中间体2与POCl3的摩尔比为1/1.2,中间体3的收率为92.45%,纯度为99.24%,效果较好,基本符合原子经济学的要求。

表3 原料摩尔比对中间体3收率和纯度的影响
2.3 4的合成
(1) 原料摩尔比
通过控制间乙炔苯胺的加入量,考察原料摩尔比对产物4收率和纯度的影响。从表4可以看出,随着间乙炔苯胺所占比例的增加,产物4的纯度呈先增加后下降趋势,但对收率影响不大。推测可能的原因是随着间乙炔苯胺用量的增加,开始呈中间体3反应完全趋势,但后来反应液中未反应的间乙炔苯胺的量越多,导致副产物增多,因此产物4的纯度呈下降趋势。因此最佳的原料摩尔比为1/1.1。

表4 原料摩尔比对4收率和纯度的影响
(2) 反应时间
由表 5可以看出,随着反应时间的增长,产物4的收率呈现出先增加后下降,最后趋于稳定的现象。反应6 h左右,产物4收率最高(91.3%)。

表5 反应时间对产物4收率和纯度的影响
设计了一条公斤级合成盐酸埃罗替尼的新路线。对关键步骤,即环合反应、氯化反应、胺化反应进行了优化。该路线具有操作简单、收率和纯度较高、对环境污染小、不需柱层析纯化等优点。
猜你喜欢
小星星·阅读100分(低年级)(2022年11期)2022-05-30 10:48:04
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
国际眼科杂志(2019年9期)2019-09-06 09:28:02
现代农业(2016年4期)2016-02-28 18:42:14
现代泌尿外科杂志(2015年11期)2015-07-26 09:02:02
应用化工(2014年1期)2014-08-16 13:34:08
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
无机化学学报(2014年12期)2014-02-28 17:34:01
化工生产与技术(2014年3期)2014-02-27 13:41:44
