
碱基编辑技术及其在昆虫中的应用和展望
2021-08-11 02:15:56王晓迪冀顺霞申晓娜刘万学万方浩张桂芬吕志创
中国生物防治学报 2021年3期
王晓迪,冀顺霞,申晓娜,刘万学,万方浩,张桂芬,吕志创
(中国农业科学院植物保护研究所/植物病虫害生物学国家重点研究室,北京 100193)
昆虫作为自然界中数量最多的生物,与农林牧的生产息息相关,科学家们也在不断地对其进行深入的研究,基因组信息是深入研究分子和遗传机制以及进化过程的重要基础。近年来越来越多的昆虫基因组被测序。据统计,2018年12月底在NCBI数据库中已有331个基因组完成组装拼接[1],截至2020年6月9日增至497种。随着昆虫基因组信息的日益完善,基因编辑技术的出现为昆虫学的研究开辟了新思路,为昆虫的基因功能研究提供了新的技术支持。经典的基因组编辑工具包括zinc-finger nucleases(ZFNs)技术[2,3]和transcription activator-like effector nucleases (TALENs)技术[4-6],其已在果蝇Drosophila、家蚕Bombyx mori、蟋蟀Gryllulus等昆虫的研究中获得成功[7-11]。随后发现了由RNA指导Cas蛋白进行DNA识别及编辑的 Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated(Cas)技术[12],并广泛应用于临床试验、动植物、细菌、真菌等研究领域[13-19]。以上三种基因编辑技术作为功能基因组研究的重要工具在昆虫的研究中已得到了应用[20,21],为今后进行昆虫功能基因组研究奠定了坚实的基础。为了防止双链断裂(double strand break,DSB)引起的潜在危害,保证基因组的完整性,近年来,一系列无DSB的单碱基编辑工具——胞嘧啶碱基编辑器(cytosine base editors,CBE)、腺嘌呤碱基编辑器(adenine base editors,ABE)和先导编辑(prime editing)已经在多种物种中被广泛开发,并对其编辑技术的精确性及广适性进行了验证。
1 CRISPR/Cas系统
CRISPR/Cas系统最初是由日本大阪大学(Osaka University)于1987年在研究大肠杆菌EscherichiacoliK12体内碱性磷酸同工酶的iap(Alkaline Phosphatase)基因的核苷酸序列时发现的,其中一段由重复短序列间隔排列组成的片段引起了研究者的注意[12]。2002年,Jansen等[22]将这段序列命名为clustered regularly interspaced short palindromic repeats (CRISPR)以此来反映这一簇有规则间隔的短回文重复序列的特征,并在研究中确定了四个CRISPR相关基因cas1、cas2、cas3和cas4,cas基因始终位于CRISPR位点附近,说明二者之间具有功能相关性。
研究证明,CRISPR基因座存在于大量细菌和古细菌中[22-26]。根据Cas基因核心元件序列的多样性,将CRISPR/Cas系统分为两类六型[27]:第一类是多亚基效应复合物,需要多个Cas蛋白相互作用[28,29],才能发挥CRISPR系统的作用,包括类型I、类型III和类型IV,前两种类型在古细菌中出现的频率较高,类型IV较罕见;第二类是单蛋白效应器,该复合物由一个单一的、大的、多结构域的蛋白质组成,CRISPR/Cas位点的组织结构更简单、更统一,其类型包括类型II、类型V、类型VI,该类系统中的Cas9、Cas12a(Cpf1)和Cas12b(C2c1)的靶向目标是DNA,Cas13a(C2c2)、Cas13b的靶向目标是RNA。Cas9基因最早是在 2007年噬菌体侵染嗜热链球菌的试验中发现的,研究发现该基因能很好地抵抗噬菌体的入侵,并证实了原核生物已进化出基于核酸的“免疫系统”,其特异性由CRISPR间隔区的含量决定,而抗性由Cas酶机制提供[30],随后CRISPR/Cas9系统便在动植物等的研究中被广泛应用。
在分子水平上,CRISPR的作用过程可以分为三个阶段:整合新的间隔物到CRISPR阵列中,表达和处理CRISPR RNAs(crRNAs),以及CRISPR干涉[31]。第一阶段,当外源质粒入侵时,识别PAM(Protospacer Adjacent motif)序列,将proto-spacer与CRISPR间隔序列同源的噬菌体基因序列[32,33]插入到CRISPR位点从而来抵抗入侵的质粒和噬菌体,进而使细胞快速适应环境中的入侵者,因此这个阶段被称为CRISPR功能的适应阶段。其获取间隔物的机制以及CRISPR如何识别噬菌体或质粒DNA是入侵性的尚未明确。第二阶段,一旦在CRISPR位点上建立了一个隔离子,它就为CRISPR通路的防御阶段提供了特异性。这个过程的关键是由重复序列和间隔序列编码的小crRNAs产生。为了进行成功的防御,该CRISPR阵列被转录生成CRISPR-RNA,该RNA作为向导RNA(gRNA),用于识别与Cas蛋白结合的核糖核酸复合物中的互补外源DNA/RNA,从而降解靶标,阻止入侵。第三阶段,在Cas蛋白复合物的参与下进行靶向和干扰入侵的噬菌体DNA序列[34-37]。通过这样的过程细胞成功抵御了噬菌体或质粒的入侵。
从2012年CRISPR/Cas9技术正式问世到现在,这一生物技术被广泛应用到人体临床试验,动植物、细菌、真菌等基因的研究上,解决了众多科学问题。在对果蝇、家蚕、埃及伊蚊Aedesaegypti等昆虫的研究中得到了充分应用[38-47]。通过在 ATP依赖的结合转运蛋白(白色)中产生一系列框移突变,证明了CRISPR/Cas介导的昆士兰实蝇Bactroceratryoni眼色基因突变,产生经典的白眼表型,为今后开发基因性菌株进行虫害防治提供了可能[48]。利用CRISPR/Cas9系统对棉铃虫Helicoverpazea基因组进行编辑,采用核糖核蛋白(ribonucleoprotein,RNP)复合物,将合成的gRNA与经过核定位信号设计的纯化Cas9核酸酶结合,从而实现对vermillion眼色基因的高效编辑,试验结果显示胚胎注射2或4 μmol/L RNP复合物时突变率高,编辑效果更好[49]。中国科学院上海植物生理学与生态研究所利用CRISPR/Cas9系统构建了亚洲玉米螟OstriniafurnacalisArgonaute1突变体(OfAgo1),揭示了OfAgo1在亚洲玉米螟角质层色素沉着中的作用[50]。研究发现,以加勒比果蝇Anaststrephasuspensa为研究对象,首先以外源性转基因多聚泛素调控的EGFP(polyubiquitin-regulated EGFP,PUb-EGFP)为靶点,通过注射Cas9蛋白,实现CRISPR/Cas9介导的基因编辑,使用单个sgRNA 进行可遗传的非同源末端连接(non-homologous end joining,NHEJ)敲除。在带有PUb-DsRed标记的转基因果蝇中,距离靶标位点2~5个碱基处发现了多个缺失变异,种系间的突变效率可达29%。同时对其内源性性别决定基因As-transformer-2(Astra-2)进行敲除,根据两性生殖形态,鉴定出G0雌虫体细胞突变的频率可达81%[51]。以CRISPR/Cas9为基础,通过卵细胞靶向肽配体(BtKV)与Cas结合注射到烟粉虱Bemisiatabaci雌性成虫卵黄中,可实现对后代基因进行有效且可遗传的编辑,为今后烟粉虱等害虫的防治提供了新的参考防治策略[52]。在不同昆虫中建立CRISPR/Cas9系统,为虫害管理提供了新的视角。2020年5月,西南大学马三垣和夏庆友团队利用广谱的piggyBac转座子系统,首次构建了适合于昆虫的CRISPR文库构建策略,并在家蚕细胞系中测试了其效果,结果显示,测试的所有位点的敲除效率都达到了 100%[53]。该研究为家蚕和其他昆虫的相关研究和应用提供潜在的靶标,并实现了一系列生物与非生物胁迫的阳性筛选,推动了昆虫基因编辑技术的研究和应用。
2 碱基编辑系统
尽管CRISPR/Cas9技术迅猛发展,日益完善,但是传统的CRISPR/Cas9系统主要是通过gRNA的引导,使得Cas9蛋白结合到与gRNA互补配对的DNA链上,对目标基因进行切割形成DNA双链断裂(DSB),诱发细胞内非同源末端连接(non-homologous end joining,NHEJ)和同源重组(homologous recombination,HR)[54,55],从而完成对基因的编辑。在编辑的过程中NHEJ与HR存在竞争,NHEJ是一种有效地创制基因敲除突变体的途径,其发生频率较高,但修复方式不够精确,常常在靶点处产生碱基的插入或缺失(insertions/deletions,indels);HR途径是一种较为精确的修复方式,但效率非常低。因此在编辑的过程中往往达不到预期的基因修饰的目的,且不可避免地会对基因组造成损伤[56,57]。
碱基编辑是基因组编辑的一种形式,它使一个碱基对在目标基因组位点上直接、不可逆地转换为另一个碱基对,而不需要双链DNA断裂(DSBs)、同源定向修复(HDR)过程或供体DNA模板[58,59]。与引入点突变的基因组编辑方法相比,碱基编辑可以更有效地进行碱基的精准插入和删除以及碱基之间的转换。碱基编辑系统(Base editor)的诞生在保护了基因组的完整性的基础上提高了编辑效率和精准度,为基因修复提供了更加方便快捷的工具,有力推进了基因组编辑的进程。目前碱基编辑系统依据融合的不同碱基修饰酶分为两类:一类是胞嘧啶碱基编辑器(cytosine base editors,CBE),另一类是腺嘌呤碱基编辑器(adenine base editors,ABE)[60]。这两类碱基编辑系统利用胞嘧啶脱氨酶或人工改造的腺嘌呤脱氨酶实现对靶标位点的精准编辑,使得C·T(G·A)或A·G(T·C)的替换更为精确[58,61,62]。
2.1 胞嘧啶碱基编辑器(cytosine base editors,CBE)
2016年,David R.Liu研究团队设计了CRISPR/Cas9和胞嘧啶脱氨酶的融合体,该融合体保留了gRNA的引导力,不诱导dsDNA断裂,并介导胞嘧啶直接转化为尿嘧啶,从而影响了C·T(或G·A)的取代,基本原理如图 1所示。由此产生的“碱基编辑器”在大约五个核苷酸(nt)的窗口内转换胞嘧啶,并能有效地纠正与人类疾病相关的各种点突变[58]。经测试发现,大鼠胞嘧啶脱氨酶(rAPOBEC1)融合到dCas9的N端,而不是C端,并表现出最高的脱氨酶活性,通过表达纯化以及体外评估,确立了第一代碱基编辑器(BE1)为rAPOBEC1-XTEN-dCas9蛋白。此后该团队将UGI与BE1的C端融合,创建了第二代碱基编辑器(BE2,apobeci-xten-dcas9-UGI),降低了细胞在修复U·G异源DNA双链反应时对碱基编辑效率的影响;为进一步提升碱基编辑效率,尝试进一步操纵细胞DNA修复来诱导对含有G真核错配修复(MMR)的未编辑链的修正,利用新合成的DNA中出现的缺口来直接移除并重建新链,从而产生了第三代碱基编辑器(BE3,APOBEC-XTEN-dCas9 (A840H) UGI)[58]。2017年David R.Liu实验室开创了四代碱基编辑器(BE4和SaBE4),它将基因编辑C·G-to-T·A之间的转化效率提高了约50%,同时与BE3相比,将不需要的副产物的频率降低了一半。将BE3、BE4、SaBE3或SaBE4融合到Gam中,结合DSBs的噬菌体Mu蛋白大大降低了碱基编辑过程中indel的形成,在大多数情况下降低到1.5%以下,并进一步提高了产品的纯度[59]。BE4、SaBE4、BE4-gam和SaBE4-gam代表了C·G-to-T·A碱基编辑的最新状态,在未来的试验研究中会被充分利用。可以说该碱基编辑技术显著促进了基因组编辑的范围和有效性。

图1 CBE系统工作原理示意图[58]Fig.1 Schematic diagram of the function in CBE system
2016年,日本神户大学 Akihiko Kondo实验室将脓链球菌Streptococcuspyogenes的 dCas9(a nuclease-deficient mutant of Cas9)和来自七鳃鳗Lampetrajaponicum的配体PmCDA1进行结合形成复合物(Target-AID),该复合物可以进行高效的靶向特异性诱变,特异性点突变主要发生在5个碱基靶标范围内的胞嘧啶上,在酵母和哺乳动物细胞中均能诱导插入和缺失(indel)。Target-AID通过脱氨酶介导的超突变,在不使用模板DNA的情况下,缩小了靶向核苷酸替换的范围,从而扩大了CRISPR/Cas9系统的基因组编辑潜力,提高了碱基编辑的准确性[63]。同年,Michael C.Bassik团队利用催化激活休眠的dCas9来招募hAID脱氨酶(hyperactive Activation induced deaminase)的变体,在有限的靶标损伤下,对内源靶标进行特异化诱变,从而产生多个不同的点突变文库,与 Cas9产生的插入和删除形成对比,此团队研发的CRISPR-X技术可用于同时突变多个基因组位置[64]。2017年,George M.Church实验室通过与ZF或TALE-DNA融合胞嘧啶脱氨酶生成可编辑的脱氨酶,实现原核细胞和真核细胞基因组位点的特异性脱氨,进一步设计和优化后可以将细胞内特定的C·G碱基对转化为T·A。大肠杆菌基因组达到高达13%的编辑频率,优化后的嵌合脱氨酶应用于人类细胞系,发现这些新的酶可以在2.5%的细胞中产生位点特异性的单核苷酸转移。与靶向核酸酶相比,转染细胞的细胞毒性降低[65]。2020年5月11日,华东师范大学李大力课题组将非序列特异性的ssDNA结合结构域(single-stranded DNA-binding domain,ssDBD)与CBE进行融合,通过筛选,发现将Rad51蛋白的ssDBD融合到APOBEC1与Cas9n之间能显著提高碱基编辑活性,同时编辑窗口也大幅增加,且hyeA3A-BE4max可在不引起血红蛋白γ基因启动子附近基因突变的情况下,特异性地实现C-to-T的转化,展示了其在动物模型中产生精确突变以此来治疗人类疾病的能力[66]。2020年5月18日,左二伟研究团队根据蛋白质结构预测了脱氨酶ssDNA结合的重要氨基酸,在保证催化活性的前提下,对APOBEC1上的ssDNA结构域相应氨基酸进行突变,筛选出了脱靶效率显著降低的CBE突变体,并在此基础上增加标签和核定位序列(FNLS),研究表明YE1-BE3-FNLS显著降低了脱靶效应并提高了编辑效率,是一种高精度、高活性单碱基编辑工具。这项研究成果有望应用于遗传病基因治疗,推动基因编辑临床化应用,在农林牧领域也具有广泛的应用前景。
自碱基编辑系统产生后,已在细菌、动物、植物物种中广泛应用[62,68,69]。胞嘧啶碱基编辑器(CBE)在基因敲除[70-77]、疾病模型[61,78-83]、疾病治疗[84-90]、畜牧生产及大动物模型构建[91-94]以及植物[95-106]中均有大量成功应用的例子,并且已经带来了很多的突破。但碱基编辑系统在无脊椎动物包括昆虫中的应用上屈指可数,至今为止,只查阅到2018年西南大学夏庆友团队将剪辑编辑系统BE3应用于家蚕中。该团队成功建立了家蚕 BE3碱基编辑系统,实现了在家蚕基因组上的特定位点 C·G-to-T·A 的替换,对Blos2和Yellow-e基因的碱基编辑效率分别为40%和51.2%,同时发现BE3最多能同时编辑Yellow-e基因的4个碱基;通过引入无义突变的方式建立家蚕 BE3敲除体系,利用该体系高效敲除家蚕外源基因mCherry和Puromycin,以及内源基因BmGAPDH和BmV-ATPaseB,其中mCherry和Puromycin编辑效率分别为66.2%和58.3%,验证了BE3作为家蚕基因敲除工具的普遍性;BE3能实现对EGFP基因多达14个C·G碱基对替换的同时编辑,且几乎没有观察到indel[107,108],该研究结果将为在其他无脊椎动物尤其昆虫中建立碱基编辑系统提供了参考范例,相信不久的将来BE3碱基编辑系统会在昆虫领域中被普遍应用。
2.2 腺嘌呤碱基编辑器(adenine base editors,ABE)
2017年David R.Liu实验室通过对腺嘌呤脱氨酶定向改造,开发了腺嘌呤碱基编辑系统,当它与催化受损的CRISPR/Cas9结合时,它可以作用于DNA。其基本原理如图2所示,即脱氨酶与Cas9切口酶(Cas9n)的N末端融合,在gRNA引导下,使非靶链中的腺苷脱氨,暴露为单链DNA(single-stranded,ssDNA),然后将A转化为肌苷(I)(A-to-I),最后,DNA聚合酶将I识别为G进行复制,互补链上原来与腺嘌呤A互补的T将会变成C,进而完成碱基置换过程[61]。经过7轮进化与改造开发了ABE7.10系统,该系统能有效地实现 A·T-to-G·C(在人类细胞中约 50%)的替换,具有很高的产品纯度(通常≥99.9%)和很低的 indels率(通常≤0.1%)。ABEs比现有的基于 Cas9核酸的方法更有效、更直接地引入点突变,比Cas9更少地诱导脱靶基因组修饰,可以在人类细胞中修正或抑制基因突变[61]。第七代 ABEs 极大地扩展了基因编辑的范围,结合CBE使得C·T、A·G、T·C和G·A之间的转换成为可能,为今后人类细胞的基因治疗以及其他动植物基因的编辑提供了更加精确、快捷的编辑工具。
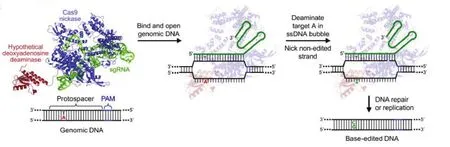
图2 ABE系统作用原理示意图[61]Fig.2 Schematic diagram of the function in ABE system[61]
ABE系统开发至今虽然并未查阅到其在昆虫中的相关研究,但却已成功应用于各种动物细胞系、人类胚胎、小鼠、大鼠、兔子、斑马鱼、水稻、小麦、拟南芥和油菜等[62,68]。例如,对水稻Oryzasativa的研究表明,应用ABE-P1编辑系统能使其基因的编辑效率达到26%[109];基于荧光示踪的rBE14系统也能在水稻上实现高效编辑[110]。Hao等[111]研究表明 ABE系统既能用于研究水稻目标氨基酸置换又能用来干扰miRNA的结合区域,同时发现应用经过修饰的sgRNA突变体会增加碱基编辑的效率。同时,研究证明了ABE和 CBE系统可以同时对水稻基因组进行定点编辑[112]。ABE系统在小麦Triticumaestivum、拟南芥Arabidopsisthaliana和甘蓝型油菜Brassicanapus上也实现了50%以上的高效率编辑[106,113,114]。
3 先导编辑(prime editing)
2019年11月,David R.Liu团队在原有的基因编辑技术的基础上开发了一种新型的基因编辑技术——先导编辑[115],此项技术相比于同源性导向的修复,提高了编辑效率以及产品纯度;相比于碱基编辑,具有互补性优势,实现了碱基之间的颠换;在已知的Cas脱靶位点上,提供了比Cas核酸酶更低的脱靶率。先导编辑技术大大扩展了基因组编辑的范围和能力,原则上可以纠正大约89%的人类已知致病性基因变异。
先导编辑主要的复合物包括Cas9酶(被修饰成仅切割DNA的一条链)、逆转录酶(以RNA为模板产生新的DNA链),此外还需要一种特殊的gRNA——pegRNA(prime editing guide RNA),这种pegRNA不但能够结合想要编辑的特定DNA区域,还会自带“修改模板”,从而在精确编辑中发挥作用。其编辑的过程如图3所示,即pegRNA将编辑信号发送到靶标处,引导Cas9切割DNA的一条链;随后反转录酶读取RNA并将相应的碱基连接到被剪切的DNA链的末端,与此同时将编辑序列从pegRNA转移到靶标DNA上;细胞中的核酸内切酶切除旧的DNA片段,将新的碱基组合到基因组中;此时靶标位点只剩下一条已编辑的链和一条未编辑的链;为解决错配问题,有利于已编辑过的DNA永久安装,一个不同的gRNA指引Prime editor去剪切未编辑的链;剪切产生的缺口提示细胞以编辑过的链为模板,重新生成新的链,最终完成编辑[116]。
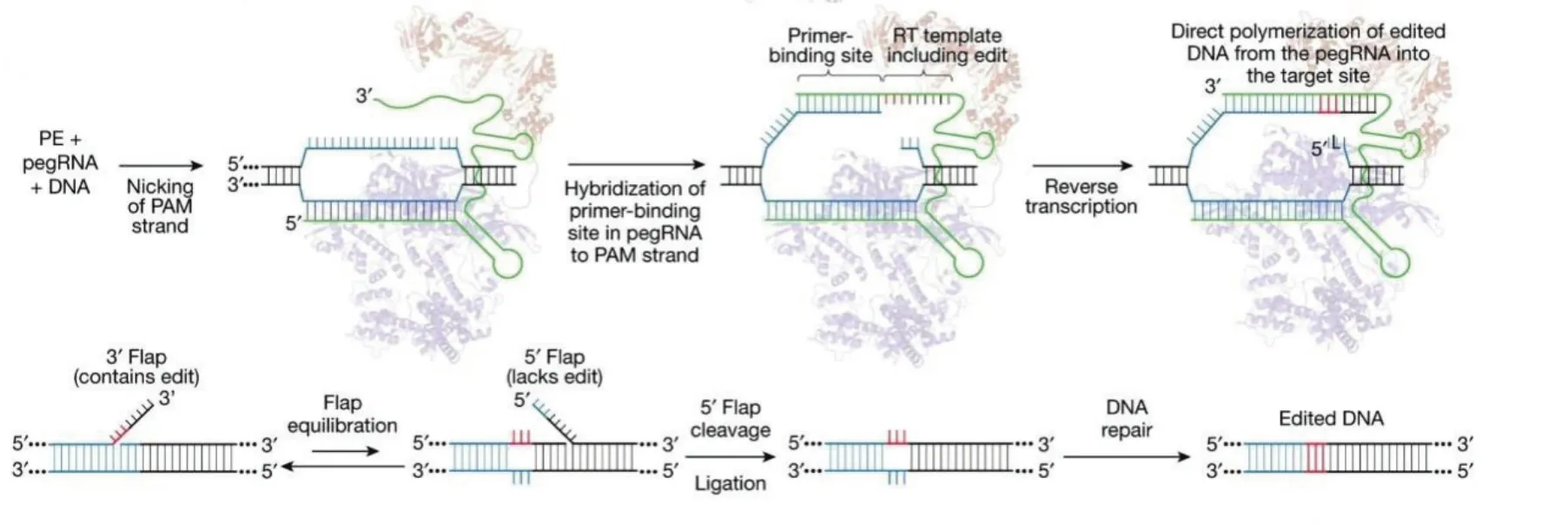
图3 先导编辑系统工作原理示意图[115]Fig.3 Schematic diagram of the function in prime editing[115]
先导编辑是一种“搜索和替换”的基因组编辑技术,它可以在不需要DSBs或供体DNA 模板的情况下,在人类细胞中介导目标碱基的插入、删除以及12种可能的碱基到碱基的转换以及它们的组合。David R.Liu团队首先用一个编码野生型M-MLV逆转录酶融合体的质粒转染HEK293T细胞,该质粒通过柔性接头到达Cas9 H840A切口酶的任一末端,另一个质粒编码pegRNA,结果显示pegRNA在8~15个碱基中的延伸导致在HEK3靶位点可检测到碱基发生颠换,当逆转录酶融合到Cas9酶的C末端时编辑效率更高,同时编辑效率也取决于PBS序列的长度,试验结果显示在HEK293T细胞中最大编辑效率可达0.7%~5.5%[115]。科学家将 M-MLV逆转录酶与 Cas9 H840A的 C端的融合命名为 PE1(Prime editor 1)。将D200N+L603W+T330P引入到M-MLV 逆转录酶中,经过筛选,在原有的基础上添加T306K和W313F,这种结合到 PE1中的五种突变逆转录酶(Cas9 H840A-M-MLV RT D200N+L603W+T330P+T306K+W313F)被命名为PE2。PE2比PE1更有效地进行了定向插入和删除,使得先导编辑的定点突变率提高了1.6~5.1倍。为进一步优化碱基编辑,使用Cas9切口酶对未编辑的链进行切割,通过在HEK293T细胞的五个基因组位点测试,产生了 PE3,在此基础上设计了含有与编码链匹配的间隔子的 sgRNAs,最大限度地减少DSB和indel的形成。总的来说,先导编辑提供了更加精确的方法进行基因编辑,且在原有的只能进行碱基转换的基础上,进行了创新性的突破,使得在基因治疗等领域实现有效、精确地将DNA序列插入到活细胞的目标位置成为可能。
2020年,研究人员通过密码子、启动子和编辑条件优化,成功地将先导编辑技术应用于水稻和小麦中,其编辑效率达到21.8%[117]。同年4月,首次报道了在人类HEK293T细胞8个位点中验证了先导编辑,并在小鼠体内证明了先导编辑系统的多功能性,这是首次报道使用先导编辑在动物体内产生靶向的碱基转换突变,这也为先导编辑在修复遗传性疾病方面的潜力提供了数据支持[118]。试验研究显示使用先导编辑技术对镰刀细胞贫血症的血红蛋白基因进行修复,有效率可达58%,仅有1.4%出现了以外的插入或缺失,同时先导编辑技术修复Tay-Sachs病的HEXA基因的有效率也达到了33%,意外插入和缺失只有0.32%,研究人员推断该技术理论上可以修复75000种已知致病性人类遗传变异的89%[119]。
4 碱基编辑系统的发展前景
2017年碱基编辑技术被Science杂志评为全球时代年度科学突破之一,2020年美国科学院公布未来10年农业发展的五大方向中的“突破性的基因组学和精准育种技术”是以基因编辑技术的发展应用作为核心技术导向,由此可见基因编辑系统的发展具有巨大的潜力。随着CRISPR/Cas技术、胞嘧啶碱基编辑技术、腺嘌呤碱基编辑技术以及最近研发的先导编辑系统等基因编辑工具的不断更新,编辑的范围从原有的低精确度,到实现单碱基的转换,再到可实现任意碱基之间的替换,不断改进的过程,indels也不断降低,让我们更加地坚信在未来的基因治疗、基因功能鉴定等方面会向着更加精准化、有效化的方向展开,并将其应用在动植物,尤其是昆虫的防治中去。碱基编辑技术在昆虫中应用,将为害虫管理提供新的研究方向,以维持生态平衡为前提对有害生物等进行有效防控,减少对农业经济的不利影响,保证农林牧等产业的稳定发展。
猜你喜欢
中国实用医药(2021年20期)2021-08-04 14:27:22
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
党的生活·党员电教与远程教育(2019年1期)2019-03-06 12:41:42
铁道通信信号(2018年7期)2018-08-29 01:17:08
中国医药指南(2018年3期)2018-03-23 09:14:25
电子设计工程(2015年4期)2015-02-27 12:04:14
食品工业科技(2012年1期)2012-11-15 02:03:32
