
急性髓性白血病预后免疫相关基因的生物信息学分析
2021-08-11 02:58:32郭凤霞赵海燕任咏慧
生命科学研究 2021年3期
郭凤霞,赵海燕,王 博,汪 梅,任咏慧,李 洁*
(1.牡丹江医学院附属红旗医院血液科,中国黑龙江牡丹江157000;2.哈尔滨医科大学,中国黑龙江哈尔滨150081)
急性髓性白血病(acute myeloid leukemia,AML)是一种以骨髓祖代细胞克隆性扩张和分化停滞为特征的骨髓恶性疾病。先前暴露于治疗性、职业性或环境性DNA损伤剂是诱因,但大多数AML病例仍然没有明确的病因。AML是成人最常见的急性白血病,其生存期较短,5年生存率仅为24%[1]。强化化疗和基因干细胞移植的治疗方法通常适用于少数年轻的患者,对于大多数老年人而言,其预后和生存率较差[2]。尽管针对AML的治疗策略在过去的几十年里不断在调整和完善,但是这些治疗策略对患者生存预后的作用仍然微乎其微[3]。同时,由于疾病的高度异质性,处于相似临床阶段的患者接受同样的治疗方案,往往会有不同的临床结局。因此,迫切需要更好的标志物来指导AML患者的预后危险分层和个性化治疗。
越来越多的证据表明免疫反应在肿瘤的发生和发展中起着重要的作用[4~5]。为了理解免疫系统在肿瘤发生、发展中的作用,人们做出了巨大的努力,肿瘤的免疫治疗取得了一定的进展。例如,众多免疫检查点被发现,并且这些免疫检查点已经成为各种肿瘤治疗的靶点。其中最受关注的免疫检查点包括程序性死亡蛋白-1(programmed death-1,PD-1)和细胞毒性T淋巴细胞相关抗原-4(cytotoxic T lymphocyte associated antigen-4,CTLA-4),这些免疫检查点在AML中也扮演着重要的角色[6~7]。肿瘤微环境中的免疫应答是各种肿瘤侵袭和进展的显著因素,其中免疫细胞类型、细胞因子以及免疫基因已经在肺癌、卵巢癌和结直肠癌等众多肿瘤中作为预后标志物被广泛研究[8~9]。然而,目前缺乏免疫相关基因(immune-related genes,IRGs)在AML预后中的相关研究,因此,深入研究免疫分子间的相互作用,有望找到鉴定AML预后的生物标志物。
在本研究中,我们确定了AML的预后IRGs,并对其进行了基因本体论(Gene Ontology,GO)、京都基因和基因组数据库(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析,以探究预后IRGs如何在AML微环境中发挥免疫作用。此外,我们还构建了由9个IRGs拟合的预后风险模型,该预后风险模型能有效地将预后不良的患者与预后较好的患者区分开来。本文研究结果将有助于指导临床医生对AML患者进行高效、精准的个体化治疗。
1 材料和方法
1.1 数据下载、预处理和预后IRGs的筛选
TCGA数据库(The Cancer Genome Atlas,https://cancergenome.nih.gov/)提供公开可用的癌症基因组数据集[10]。ImmPort(The Immunology Database and Analysis Portal,https://www.immport.org/home)是一个收集参与免疫相关过程的IRGs的数据库[11]。首先,我们从TCGA数据库下载AML患者的基因表达谱数据以及相应的临床信息表达矩阵。然后,应用R语言对原始表达矩阵进行背景校正、数据归一化处理,并从ImmPort数据库获取IRGs。随后,从所有基因表达矩阵中提取IRGs的表达矩阵,并对其进行单变量Cox回归生存分析,筛选预后IRGs,筛选标准为P<0.05。
1.2 预后IRGs的GO和KEGG分析
GO是注释基因及其产物的重要方法和工具[12],有利于生物数据的整合和利用,包括分子功能(molecular function,MF)、生物过程(biological process,BP)和细胞组分(cellular component,CC)3个部分。KEGG是一种整合基因组学、化学和系统功能信息的数据库资源,可提供已知的生物代谢信号通路[13]。我们使用clusterProfiler包[14]对预后IRGs进行GO、KEGG富集分析,显著性基因富集的标准为P<0.05。
1.3 预后IRGs的蛋白质互作网络构建、关键预后IRGs的筛选及其功能相似性分析
STRING(Search Tool for the Retrieval of Interacting Genes,http://www.string-db.org/)是评估蛋白质-蛋白质相互作用(protein-protein interaction,PPI)的生物学工具,可以对疾病发生或发展机制进行深入了解[15]。我们使用STRING(version 11.0)在线软件和Cytoscape软件[16]进行PPI网络构建,使用插件cytoHubba[17]进行关键预后IRGs的筛选,筛选标准:MCC(maximal clique centrality)算法得分前10的基因。基于基因注释GO术语的语义相似性,我们应用GOSemSim包[18]计算得出10个关键预后IRGs之间分子功能及细胞定位的关系强度,并使用功能相似性的平均值对10个关键预后IRGs进行排序,结果由ggplot2包[19]可视化。
1.4 预后IRGs的最优模型构建、评价以及共识基因风险因子和功能相似性分析
Lasso回归有助于选择合适的变量,以简化最终模型并避免过度拟合[20]。为了构建IRGs的预后最优模型,我们使用glmnet软件包(https://CRAN.R-project.org/package=glmnet)对预后IRGs进行迭代Lasso回归分析,统计1 000次Lasso回归后的基因频率,大于100次的基因被认为是1 000次Lasso回归后的共识基因,然后对共识基因进行多变量Cox回归分析,并基于共识基因表达水平构建AML患者的免疫基因预后模型。利用共识基因表达量和多变量Cox回归系数的线性组合构建一个风险评分值,对每组患者根据风险评分由低到高进行排序,中位风险评分值被用作AML患者高、低风险组的截取值。使用时间依赖的受试者操作特征(receiver operator characteristic,ROC)曲线分析[21]验证模型的诊断效能;通过Kaplan-Meier曲线检验评估高、低风险组之间的生存差异。使用风险因子关联图展示共识基因与生存的关系,应用GOSemSim包计算得出共识基因之间分子功能及细胞定位的关系强度,并使用功能相似性的平均值对共识基因进行排序,结果由ggplot2包可视化。
2 结果
2.1 预后IRGs的筛选及其GO和KEGG富集分析
通过对ImmPort数据库获取的1 900个IRGs进行单变量Cox回归生存分析,获得302个预后IRGs。GO功能注释表明,预后IRGs涉及细胞趋化反应、白细胞黏附和先天性免疫调节等生物过程;参与细胞质膜外侧、蛋白酶体复合物等细胞组分;涉及的分子功能有受体配体活性、生长因子活性、细胞因子活性和细胞因子受体结合等(图1A)。KEGG通路富集结果显示,预后IRGs主要富集于细胞因子-细胞因子受体相互作用、自然杀伤细胞介导的细胞毒性、趋化因子信号途径以及JAKSTAT等信号通路(图1B)。
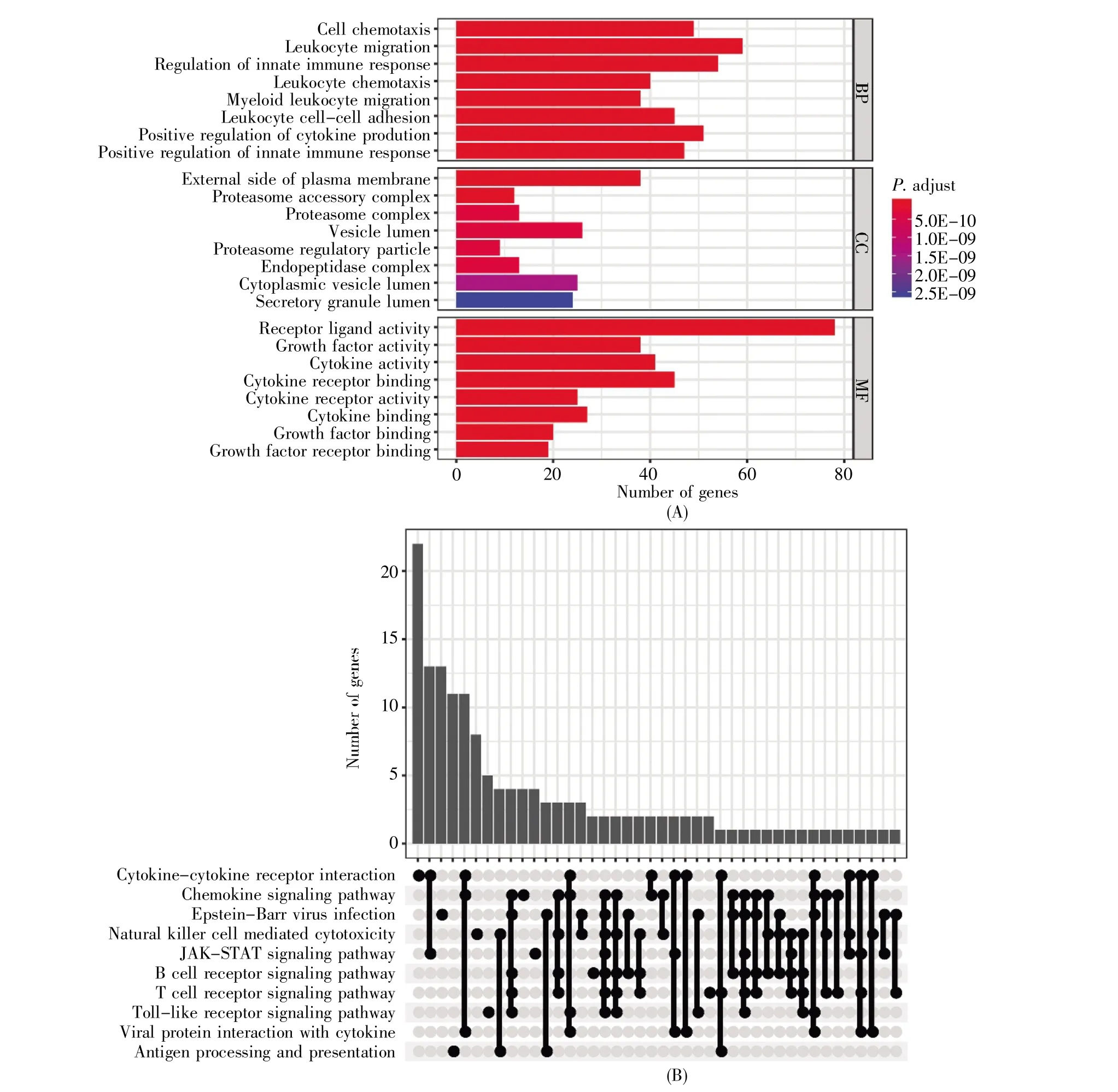
图1 预后IRGs的GO和KEGG通路富集分析(A)预后IRGs的GO分析;(B)预后IRGs的KEGG信号通路富集分析。Fig.1 Enrichment analysis of GO and KEGG pathways in prognostic IRGs(A)GO enrichment analysis for prognostic IRGs;(B)Enrichment analysis of KEGG signaling pathway for prognostic IRGs.
2.2 预后IRGs的PPI网络分析及关键预后IRGs的功能相似性分析
通过STRING在线数据库构建预后IRGs的PPI网络,并使用Cytoscape将其可视化,结果如图2A所示。经cytoHubba筛选,共获得10个关键预后 IRGs,分别为 NFKB1、PSMB8、NFKBIA、PSMC3、PSMD4、PSMD7、PSMD2、PSMC4、PSME2 和PSME3(图2B)。为进一步鉴定关键预后IRGs间相互作用的关系强度,我们根据它们之间的平均功能相似性进行排序,结果显示PSME3、PSMD7、PSME2、PSMD4、PSMC3 和 PSMD2 是相互作用关系最为紧密的基因(图2C)。
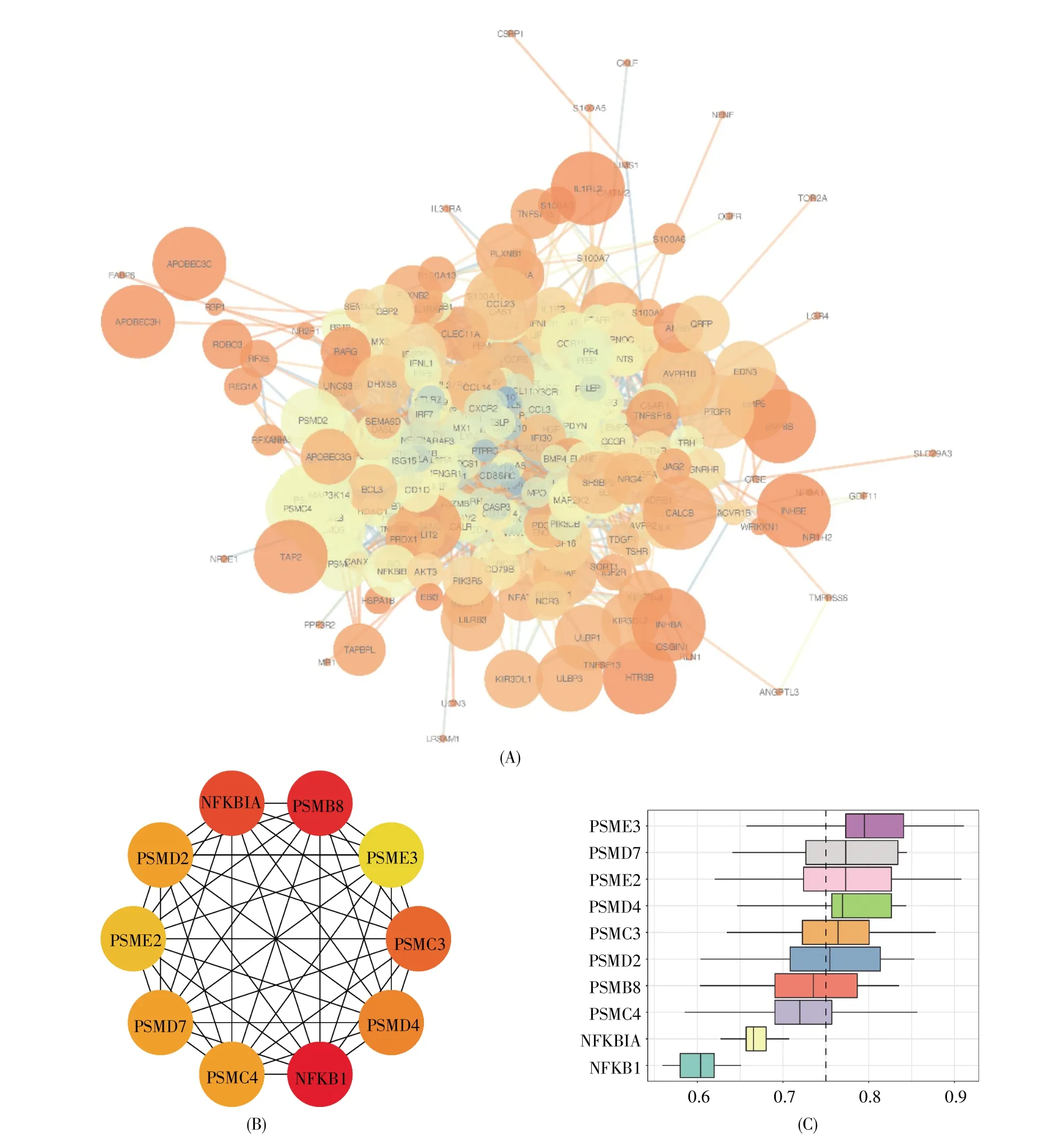
图2 预后IRGs的PPI网络、关键预后IRGs及其功能相似性分析(A)PPI网络分析图。节点大小表示聚类系数,节点颜色表示与此节点相互作用的节点个数,颜色越深,个数越多,连线粗细表示综合得分,连线颜色表示共表达强度;(B)关键预后IRGs示意图。颜色越红越深代表富集分数越高,颜色越黄越浅代表富集分数越低;(C)关键预后IRGs的功能相似性分析。将功能相似的分布概括为箱形图,箱形代表50%的相似性,上下边界显示第75和第25百分位数,方框中的线条表示功能相似性的平均值,具有较高平均功能相似性(截取值=0.75)的基因被认为是关键IRGs相互作用中的中心基因,虚线表示截取值。Fig.2 PPI network,key IRGs and their functional similarity analysis in prognostic IRGs(A)PPI network analysis diagram.The size of a node represents the clustering coefficient,and its color represents the number of nodes interacting with the node.The darker the color,the more interacting nodes there are.The thickness of a line represents the comprehensive score,and its color represents the co-expression intensity;(B)Schematic diagram of key prognostic IRGs.The darker the red color,the higher the enrichment score,and the lighter the yellow color,the lower the enrichment score;(C)Functional similarity analysis of key prognostic IRGs.The distribution of functional similarity is summarized as a boxplot.The box represents 50%similarity.The upper and lower boundaries show the 75th and 25th percentiles,respectively.The lines in the box represent the average value of functional similarity.The genes with higher average functional similarity(cutoff value=0.75)are considered as the central genes in the interaction of key IRGs.The dotted line represents the cutoff value.
2.3 预后IRGs最优风险模型构建、评价以及共识基因风险因子和功能相似性分析
迭代Lasso回归分析显示,当9个共识基因(ANGPTL3、CALCRL、CCL22、ULBP3、HGF、IL2RA、PLXNB1、RAC2、KIR2DL4)进行拟合时,诊断效能最高,为最优模型(图3A)。ROC曲线结果显示,最优模型在诊断AML高低风险预后方面准确性较高(AUC=0.91)(图3B)。Kaplan-Meier生存分析显示,高风险组患者的生存率明显差于低风险组(P<0.001)(图3C)。风险因子关联图显示,高风险病人组倾向于表达高水平的风险免疫基因(CALCRL、CCL22、ULBP3、IL2RA、RAC2、KIR2DL4), 而低危组病人则更倾向于表达高水平的保护性免疫基因(ANGPTL3、HGF、PLXNB1)(图 4A)。此外,功能相似性结果显示,PLXNB1、KIR2DL4、IL2RA、CALCRL和HGF是相互作用关系最为紧密的共识基因(图 4B)。
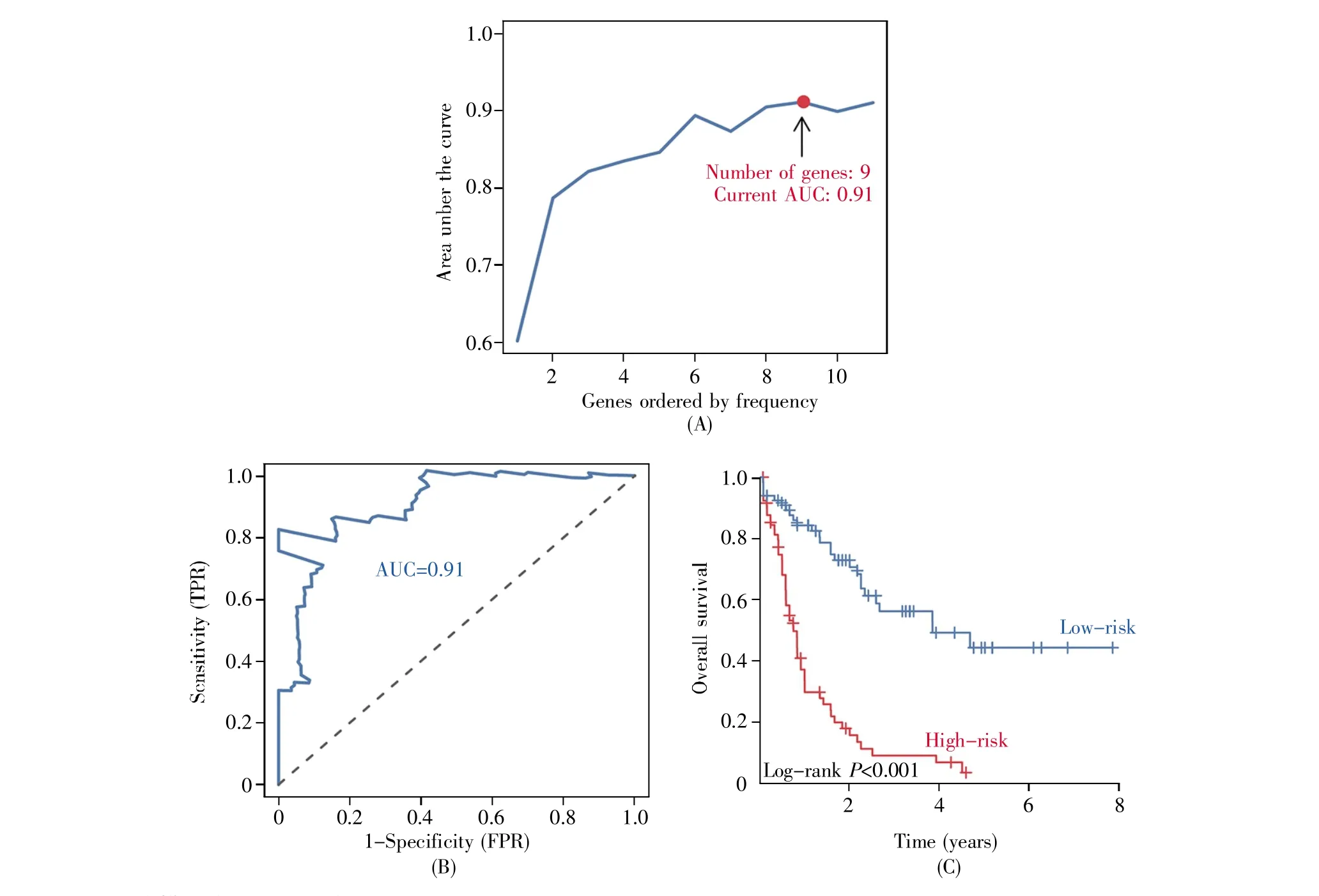
图3 最优模型构建以及评价(A)共识基因最优风险模型的筛选;(B)共识基因拟合的时间依赖的ROC曲线,横坐标代表特异度,纵坐标代表灵敏度;(C)风险评分值预测的AML患者生存曲线,横坐标代表生存时间,纵坐标代表生存率。Fig.3 Optimal model construction and evaluation(A)Selection of the optimal risk model of consensus genes;(B)The time-dependent ROC curve of consensus gene fitting.The abscissa represents specificity,and the ordinate represents sensitivity;(C)The survival curve of AML patients predicted by risk score.The abscissa represents the survival time,and the ordinate represents the survival rate.
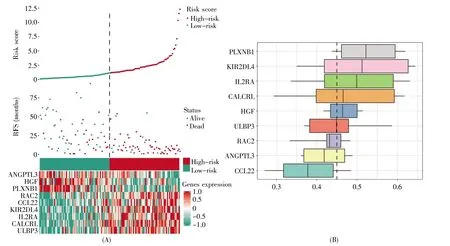
图4 共识基因风险因子关联和功能相似性分析(A)风险因子关联图;(B)共识基因的功能相似性分析。将功能相似的分布概括为箱形图,箱形代表50%的相似性,上下边界显示第75和第25百分位数,方框中的线条表示功能相似性的平均值,具有较高平均功能相似性(截取值=0.45)的基因被认为是共识基因相互作用中的中心基因,虚线表示截取值。Fig.4 Analysis of risk factor association and functional similarity of consensus genes(A)Risk factor correlation chart;(B)Functional similarity analysis of consensus genes.The distribution of functional similarity is summarized as a boxplot.The box represents 50%similarity.The upper and lower boundaries show the 75th and 25th percentiles,respectively.The line in a box represents the average value of functional similarity.Genes with higher average functional similarity(cutoff value=0.45)are considered as the central genes in the interaction of consensus genes.The dotted line represents the cutoff value.
3 讨论
AML的预后较差,年轻和老年患者都有很高的化疗耐药复发风险,需要替代性和靶向性药物来提高其5年生存率[2]。目前,AML的治疗方法主要包括化疗和分子靶向治疗,如FMS样酪氨酸激酶 3(FMS-like tyrosine kinase 3,FLT3)抑制剂、IDH[isocitrate dehydrogenase(NADP+)]抑制剂和单克隆抗体等[22],尽管治疗方法众多,但AML的预后仍旧较差。高通量基因组筛选方法和计算机辅助技术可用于预测与疾病发生有关的突变分子,辅助设计新的靶向药物[23]。因此,通过机器学习的方法构建AML患者的预后模型将有助于指导临床治疗和预后预测。免疫系统在AML微环境中发挥着重要作用,免疫紊乱是肿瘤的一个显著特征,也被认为是促进肿瘤发生、发展的决定性因素[24]。深入研究免疫基因间的相互作用,可能为预测AML患者预后提供新方向。
本研究共筛选出302个预后IRGs,为了解这些预后IRGs在AML微环境中发挥的作用,我们对其进行了GO和KEGG通路富集分析。GO功能注释结果表明,预后IRGs主要涉及细胞趋化反应、白细胞黏附和先天性免疫调节等生物过程。研究表明,细胞趋化反应[25]、白细胞黏附[26]以及先天性免疫调节[27]等在AML微环境中功能失调,减少了对AML细胞的免疫应答作用。KEGG通路富集结果显示,预后IRGs主要富集于细胞因子-细胞因子受体相互作用、自然杀伤细胞介导的细胞毒性、趋化因子信号途径以及JAK-STAT信号通路等。细胞因子-细胞因子受体相互作用信号通路[28]、趋化因子信号通路[29]、JAK-STAT信号通路[30]在AML中已经被广泛研究,这些信号通路的活化与AML生长和转移过程密切相关,机制较为明确。自然杀伤细胞介导的细胞毒性在宿主抗癌防御中起着重要作用[31]。由此我们可知,免疫反应在AML微环境中是把双刃剑,一方面利于肿瘤的发生和发展,另一方面又起到抗癌防御作用。
本研究还构建了预后IRGs所编码蛋白质间的相互作用网络,将MCC算法得分前10的基因作为关键预后IRGs,其分别为NFKB1、PSMB8、NFKBIA、PSMC3、PSMD4、PSMD7、PSMD2、PSMC4、PSME2和PSME3。值得注意的是,以上基因所编码的蛋白质大多数都是核因子κB(nuclear factor-κB,NF-κB)通路的关键分子和蛋白酶体复合物。NF-κB是一种存在于所有细胞类型中的快速作用的原代转录因子,在调节免疫反应中起关键作用,可被多种细胞内和细胞外刺激激活,如细胞因子、氧化自由基、紫外线、细菌或病毒产物。激活的NF-κB转运到细胞核,刺激参与多种生物学功能的基因表达。NF-κB的不适当激活与许多炎症性疾病有关,而NF-κB的持续抑制导致免疫细胞发育不适或细胞生长延迟。现有报道显示,PSMB8[32]、NFKB1[33]、NFKBIA[34]、PSMC3 和 PSMC4[35]参与 AML 的发生、血管生成和转移等过程,在AML中已被广泛研究;而 PSMD2、PSMD4、PSMD7、PSME2 和 PSME3 在AML中尚未被研究。PSME(proteasome activator subunit)家族编码的26S蛋白酶体是一种多催化蛋白酶复合物,由两个复合物、20S核心和19S调节因子组成,蛋白酶体以高浓度分布于真核细胞中。近年来,大量研究表明,其与结直肠癌[36]、胃癌[37]和乳腺癌[38]等多种肿瘤的生长、血管生成、转移及存活等过程密切相关。功能相似性分析结果发现,PSME3、PSMD7、PSME2、PSMD4 和 PSMC3 是相互作用关系最为紧密的基因,这进一步说明PSME3、PSMD7、PSME2、PSMD4 和 PSMC3 在AML的发生和发展中起着至关重要的作用,具体作用机制值得深入探讨。
最重要的是,本研究鉴定了一个可预测AML患者预后的由9个共识基因(ANGPTL3、CALCRL、CCL22、ULBP3、HGF、IL2RA、PLXNB1、RAC2、KIR2DL4)组成的最优风险模型。分析结果显示,该模型具有很高的诊断预后风险效能(AUC=0.91),与生存率明显相关,表明该拟合可以作为一种预测AML患者预后的可靠工具。共识基因功能相似性分析结果表明,PLXNB1、KIR2DL4和IL2RA在预测AML预后中起着重要的作用。PLXNB1(plexin B1)是一种蛋白质编码基因,与PLXNB1相关的疾病包括乳腺癌和Walker-Warburg综合征。早在2006年,Kreuter等[39]的研究就表明其与AML的预后密切相关。杀伤细胞免疫球蛋白样受体(killer cell immunoglobulin-like receptors,KIRs)是由自然杀伤细胞和T细胞亚群表达的跨膜糖蛋白,在调节免疫应答中起重要作用。KIR2DL4基因所编码的蛋白质为杀伤细胞免疫球蛋白样受体2DL4,研究表明其与AML患者的预后同样密切相关[40]。白细胞介素-2受体α(interleukin-2 receptor α,IL2RA)和 β (IL2RB)链与普通 γ 链(IL2RG)共同构成高亲和力IL2受体。同二聚α链(IL2RA)产生低亲和力受体,而同二聚β链(IL2RB)产生中等亲和力受体。可溶性IL2RA通常是一种完整的膜蛋白,已被分离并鉴定为细胞外蛋白质水解的产物。Sadras等[41]在高风险的AML病人中发现,MUC4、GPR110和IL2RA/CD25的表达水平显著升高,可能是潜在的治疗靶点。以上分析进一步证明,我们所构建的最优模型对AML患者具有潜在的预后能力,证实了我们结果的可靠性。
在本研究中,我们通过迭代Lasso回归和机器学习的方法共鉴定出9个IRGs,并且将它们作为一个整体来预测AML患者的预后。虽然之前也有大量的AML预后分子被开发,但是相比于单基因预后的研究,多个基因的拟合能够很好地避免因个体异质性引起的差异性,因此这种预后模型具有更高的诊断价值。然而,我们的研究也有一定的局限性。一方面,尽管我们使用了包含较为完整的AML患者临床信息的TCGA数据库,但是该数据库缺乏对照组样本数据;另一方面,目前缺乏足够的数据集来进一步验证我们此次分析的结果。
综上可知,我们在免疫基因层面对AML进行了深入的分析,筛选出了10个关键预后IRGs,并开发了由9个IRGs构建的风险模型。该模型可以准确预测AML患者的预后,且有可能作为预测AML预后的有效手段应用于临床。
猜你喜欢
数学物理学报(2022年5期)2022-10-09 08:56:44
现代临床医学(2021年4期)2021-07-31 07:55:54
小学生学习指导(当代教科研)(2021年6期)2021-05-23 13:24:38
马克思主义哲学研究(2020年1期)2020-11-26 07:25:48
河北画报(2020年8期)2020-10-27 02:54:20
人大建设(2019年12期)2019-11-18 12:11:06
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:37
医学研究杂志(2015年12期)2015-06-10 06:57:46
发明与创新(2015年17期)2015-02-27 10:38:59
