
水稻CRISPR/Cas12a系统的优化及其介导的腺嘌呤 碱基编辑器的建立
2021-08-11 02:46:06王敬文严芳柳浪周雪平王道文周焕斌
生物技术通报 2021年6期
王敬文 严芳 柳浪 周雪平,2 王道文 周焕斌,5
(1. 中国农业科学院植物保护研究所 植物病虫害生物学国家重点实验室,北京 100193;2. 浙江大学农业与生物技术学院,杭州 310058; 3. 中国科学院遗传与发育生物学研究所 植物细胞与染色体工程国家重点实验室,北京 100101;4. 河南农业大学农学院,郑州 450002; 5. 农业农村部桂林作物有害生物科学观测实验站,桂林 541399)
植物基因功能和分子育种等研究的前提是突变体材料的获得。自然突变、人工诱变等方法存在效率低、周期长、难操作等缺点,因此,利用序列特异性核酸酶在特定基因位点产生 DNA 双链断裂、通过非同源末端连接和同源重组修复在靶基因位点引入突变的基因组编辑技术得到迅速发展。现有的基因编辑技术根据核酸酶的不同,主要包含锌指核酸酶(ZFNs)、类转录激活效应蛋白核酸酶(TALENs)和规律成簇的间隔短回文重复序列及其相关蛋白(CRISPR/Cas),其中CRISPR/Cas系统具有操作简单、效率高、成本低等优点,受到广大研究人员的青睐。CRISPR/Cas来自原核生物(如细菌和古细菌)的免疫系统,可以选择性地靶向并破坏外源DNA,主要由Cas基因和CRISPR基因座(富含AT碱基的前导序列(leader)、涵盖回文序列的20-50 bp的重复序列(repeat)和从外源捕获的间隔序列(spacer)组成。基于Cas基因序列的差异、重复序列以及Cas簇的组成不同,Cas系统划分为2大类6种类型(I- VI)[1-3]。目前,应用较为广泛的 Cas9和Cas12系统都属于2类。
CRISPR/Cas9 作为研究最为广泛的基因编辑系统,仅需要由crRNA和tracrRNA组成的sgRNA和单体Cas9蛋白就能通过HNH和RuvC两大功能域实现对外源DNA 的切割[4],已成功在人类细胞、果蝇、水稻、拟南芥、小麦等中实现基因编辑。目前,研究报道表明通过挖掘Cas9的同源蛋白(如ScCas9和SaCas9等)和Cas9变体(xCas9和SpCas9-NG,SpRY)可对CRISPR/Cas9系统实现优化升级,提高其编辑效率和缩小PAM序列的限制性。尽管如此,该系统仍存在编辑位点受限、脱靶情况较多等缺陷。
CRISPR/Cas12a是继CRISPR/Cas9之后被开发用于基因组编辑的CRISPR/Cas系统。与Cas9蛋白不同,Cas12蛋白具有RNase和DNase活性,该系统可以同时完成crRNA的加工和其后对靶位点的切割[5]。Cas12a特异性识别富含“T”的PAM,且在PAM序列下游实现DNA双链切割,形成黏性末端[6];特别是Cas12a可以利用自身的核酶功能域同时对多个crRNA的表达阵列剪切,实现多基因编辑[7]。且Cas12a具有更高的精准度,其脱靶率要低于Cas9蛋白[8]。基于其自身的优点,CRISPR/Cas12a系统已成功应用到烟草、拟南芥、大豆、玉米、水稻等植物基因组编辑中[9-14]。目前,仅报道了Francisella novicida Cas12a(FnCas12a)、Acidaminococcus sp.BV3L6 Cas12a(AsCas12a) 与Lachnospiraceae bacterium ND2006 Cas12a(LbCas12a),且Cas12a的编辑活性受到温度等因素的影响[15]。因此,鉴定新的Cas12a蛋白或对已有的CRISPR/Cas12a系统进行优化非常重要。
基于CRISPR/Cas系统开发的单碱基编辑技术可实现基因组中靶碱基的精准替换,而这一过程并不依赖于双链DNA断裂和外源DNA供体[5]。单碱基编辑系统中的胞嘧啶碱基编辑器(CBE)和腺嘌呤碱基编辑器(ABE)应用最为广泛。ABE通过脱氨作用将靶区域的腺苷变成肌苷,从而在DNA修复过程中完成A·T到 G·C的置换。到目前为止,所有可用的ABE均来自TadA7.10,并且在植物中不同基因组位点之间显示出明显不同的编辑效率,具有典型的位点依赖性。这些问题限制了ABE的应用范围。近日,魏鹏程团队利用大肠杆菌腺嘌呤脱氨酶(TadA8e)与CRISPR系统融合构建的ABE能实现稳定高效的A>G替换的研究[16],丰富了植物基因编辑系统。目前,在水稻中还未有关于CRISPR/Cas12a介导的单碱基编辑的报道。然而针对富含AT的基因序列,CRISPR/Cas12a可提供的靶位点更多,更具优势。
因此,建立和优化基于CRISPR/Cas12a的基因组编辑工具可提高编辑效率,弥补CRISPR/Cas9系统的缺陷和不足。鉴于此,本研究通过比较不同来源的Cas12a蛋白的核酸酶活性,并对其crRNA构型进行优化,在水稻中建立高效的基因组编辑系统。并利用TadA8e开发了一套识别TTTV为PAM的新型腺嘌呤碱基编辑器rBE58,在水稻基因组中实现了A>G替换。
1 材料与方法
1.1 材料
1.1.1 植物材料 本研究中所用的水稻材料是自然条件下在稻田中种植的早熟水稻粳稻品种Kitaake(Oryza sativa ssp. geng),从健康的植株中收获的未成熟种子用于水稻转化,收获成熟的种子用于水稻原生质体实验,种子使用前用50%的84消毒液进行表面消毒。实验时,水稻材料均是在75%相对湿度、30℃ 12 h光照和25℃ 12 h黑暗条件的温室中培养。
1.1.2 菌株、质粒、试剂及引物 本研究所用的菌株和质粒见表1。潮霉素、乙酰丁香酮、氨苄青霉素、羧苄青霉素、硫酸卡那霉素、利福平和2-(N-吗啉代)乙烷磺酸购自上海翊圣生物科技公司。酵母粉、胰蛋白胨、细菌用琼脂粉、植物组培用琼脂粉、MS培养基、2,4-二氯苯氧乙酸、激动素、萘乙酸、6苄基腺嘌呤、PEG4000购自北京索莱宝公司。十六烷基三甲基溴化铵、β-巯基乙醇、琼脂糖、溴化乙锭、Axygen质粒DNA小量试剂盒、Axygen DNA凝胶回收试剂盒购自北京冰达生物科技公司。NaCl、三氯甲烷、异丙醇、乙醇、乙二胺四乙酸、Tris、甘露醇、山梨醇和蔗糖等常用试剂购自国药集团,为分析纯。各种限制性内切酶、T4 DNA Ligase、ATP、T4 polynucleotide kinase、FastAP Thermosensitive Alkaline Phosphatase等 购 自Thermo Scientific。BsaI购自NEB。PCR扩增用的高保真酶KOD Plus购自TOYOBO,Gateway® LR ClonaseTMII Enzyme Mix购自Invitrogen。实验中用到的引物的合成及测序由生工生物工程(上海)股份有限公司完成。

表1 实验中涉及的质粒和菌株Table 1 Plasmids and strains involved in this study
1.2 方法
1.2.1 (1)crRNA的设计及打靶载体的构建 LbCas-12a-crRNA表达盒利用玉米泛素化启动子驱动具有核酸酶活性的HH核酶和HDV核酶单元[10]、CRISPR/LbCas12a系统的crRNA 靶序列表达,至Nos终止子终止,该表达盒序列交由生工生物工程(上海)股份有限公司合成。然后用引物Ubi-F3/Nos-R3扩增上述合成的LbCas12a-crRNA表达盒,与用NcoI/EcoRV双酶切后的pENTR4[17]载体连接,经酶切验证及测序,最终得到正确质粒,命名为pHZ11(图1-B)。用类似的方法构建pHZ4(图1-B)。由公司合成AsCas12a-crRNA片段,将合成的AsCas12a-crRNA片段和pHZ11用SpeI/EcoRV酶切后连接,将pHZ11中的LbCas12a-crRNA替换成AsCas12a-crRNA,命名为pHZ4。本研究所选用的crRNA序列均由CRISPR-GE(http://skl.scau.edu.cn/targetdesign/)设计得出,设计原则为:PAM为TTTV,crRNA长度23 nt。互补的引物对在磷酸化后通过热处理退火反应的方式结合成双链,然后插入BsaI酶切后的pHZ11或pHZ4载体。
(2)Cas12a表达载体的构建 首先对AsCas12a和LbCas12a基因按照水稻密码子使用偏好性进行了密码子优化后委托生工生物工程(上海)股份有限公司合成,再通过BamHI和BcuI酶切将合成的基因片段AsCas12a与LbCas12克隆至瞬时表达载体pUC19∶rBE3与双元表达载体pUbi∶rBE3[18]中,替换原来的rBE3基因,得到pUC19∶AsCas12a、pUC19∶LbCas12a、pUbi∶AsCas12a和pUbi∶ LbCas12a(图1-A)。在pUC19中,由CaMV 35S启动子驱动Cas12a表达,用于水稻原生质体瞬时表达检测Cas12a的编辑活性;在双元载体中,由玉米泛素蛋白启动子(Ubi-p)驱动Cas12a表达,可用于水稻稳定的遗传转化获得基因编辑植株。
(3)水稻腺嘌呤碱基编辑器pUbi∶rBE58载体的构建 为了在CRISPR/LbCas12a系统中优化腺嘌呤碱基编辑器,我们按照水稻密码子使用的偏好性将TadA8e基因与催化失活的LbCas12a(H832A)基因(dLbCas12a)的5′末端融合在一起优化后交由生工生物工程(上海)股份有限公司(命名为rBE58),然后利用BamHI和BcuI对合成的rBE58基因片段和实验室原有pUbi∶rBE3[18]双元载体酶切,形成可用于水稻遗传转化的载体pUbi∶rBE58 (图1-C)。
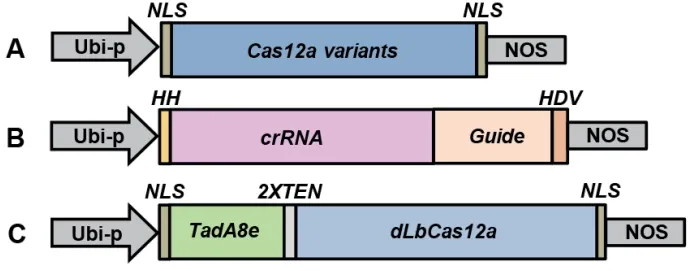
图1 载体示意图Fig.1 Schematics of gene editing constructs
在水稻原生质体中,crRNA表达框与pUC19∶Cas12a共同转化完成瞬时表达;而在水稻转化体系中,先利用AatII对pHZ11(或pHZ4)-crRNA进行酶切,通过Gateway的LR反应将含不同crRNA的质粒pHZ11(或pHZ4)-crRNA插入双元载体pUbi∶Cas12a或pUbi∶rBE58中得到pUbi∶Cas12a-crRNA或pUbi∶rBE58-crRNA,最终的载体转入农杆菌EHA105即可进行侵染水稻愈伤。
1.2.2 原生质体转化 使用之前报道的方法进行原生质体的分离和PEG介导的转染[19]。转染表达Cas蛋白和crRNA(20 μg)的质粒DNA混合物,然后将转染的原生质体在28℃下孵育48 h,收集原生质体后提取基因组DNA,用于限制性酶切验证或测序。
1.2.3 农杆菌转化 通过电穿孔将上述得到的pUbi∶Cas12a-crRNA或pUbi∶rBE58-crRNA载 体转入农杆菌EHA105。按照先前报道的方法,使用剥壳后的Kitaake种子诱导水稻愈伤组织进行遗传 转化[20]。
1.2.4 编辑效率检测 采用十六烷基三甲基溴化铵(CTAB)方法提取水稻原生质体和转基因材料的基因组DNA。
为了检测原生质体中是否发生预期突变,将提取的原生质体基因组DNA用BamHI酶解6 h,然后使用高保真酶KOD Plus和靶位点对应的引物进行PCR扩增,扩增产物用BamHI酶解3 h,并在琼脂糖凝胶上分离,富集未切割的条带,最后连接到pGEM-T载体,挑单克隆进行Sanger测序鉴定突变的序列。
为了检测通过水稻遗传转化获得的转基因材料的编辑效率,将提取的基因组DNA用包含预期编辑位点的基因特异性引物进行PCR扩增,通过Sanger测序验证突变的序列。突变愈伤(植株)数与所检测的转基因愈伤(植株)总数的比例,即为转基因愈伤的编辑效率。
2 结果
2.1 CRISPR/Cas12a编辑活性的检测
首先,利用水稻原生质体实验对构建的CRISPR/Cas12a系统进行编辑活性检测。将分别表达Cas蛋白的pUC19载体和带有靶位点的crRNA载体通过PEG介导水稻原生质体转化导入水稻细胞中进行共表达,转染48 h后收集并提取原生质体基因组DNA,通过BamHI酶切消解去除未发生编辑的靶位点,富集携带有成功编辑(即BamHI酶切位点被破坏)的靶位点区域片段,最后对基因组DNA靶位点区域进行PCR扩增和Sanger测序。结果显示在crRNA为23 nt时,LbCas12a和AsCas12a处 理的样品都成功扩增出目的片段,且在靶位点引起的突变基本都在-6--12 bp之间(图2),上述结果证明以TTTV为PAM的CRISPR/LbCas12a和CRISPR/AsCas12a在水稻中都具有编辑活性。

图2 利用水稻原生质体检测CRISPR/AsCas12a和CRISPR/LbCas12a的核酸酶活性Fig.2 Detection of CRISPR/LbCas12a in rice protoplasts and the nuclease activity of CRISPR/AsCas12a
2.2 crRNA构型对Cas12a编辑活性的影响
Moon等[21]2018年研究表明,在20 nt crRNA 的3′添加UUUUAUUUU(U4AU4)序列可转录形成最佳构型的crRNA,能增强AsCas12a在人类细胞中编辑效率。因此,为了建立高效的CRISPR/Cas12a介导的水稻编辑系统,我们探索了crRNA长度和crRNA 3′端富含尿嘧啶(U)对编辑效率的影响。本研究设计了4种crRNA,分别是:(1)20 nt,(2)20 nt+U4AU4(20+9 nt),(3)23 nt,(4)23 nt+U4AU4(23+9 nt),选择3个水稻内源基因(OsCERK1,OsCPK13和OsCPK17)进行打靶实验(表2)。通过PCR扩增和Sanger测序对获得的独立转基因水稻品系进行基因型分析。结果显示:LbCas12a在3个靶位点处均显示出强大的活性(图3),当测试 的crRNA为23 nt时,在 OsCERK1、OsCPK13和OsCPK17中编辑效率分别为77.78%、84.21%和86.67%,并且大多数突变类型为双等位基因突变;当使用23 nt+U4AU4为crRNA时,LbCas12a的核酸酶活性大大降低;虽然与20 nt相比,使用20 nt+U4AU4为crRNA可使在OsCERK1和OsCPK17位点的编辑效率有所提高,但还是低于23 nt crRNA的编辑效率(图3)。相较于LbCas12a,AsCas12a仅在OsCERK1和OsCPK13中检测到突变,编辑效率范围为0至46.43%,其中,使用20 nt为crRNA、靶向OsCPK13时编辑效率最高。综上所述,在水稻中LbCas12a的编辑活性明显优于AsCas12a,并且LbCas12a使用23 nt为 crRNA编辑效率最高,同时通过在靶序列的3′端添加U4AU4并不能提高AsCas12a和LbCas12a的编辑活性。
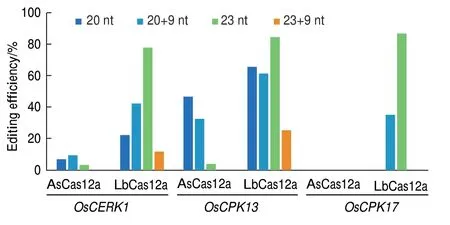
图3 不同crRNA构型对AsCas12a和LbCas12a核酸酶活性的比较Fig.3 Comparison of nuclease activities of AsCas12a and LbCas12a with different crRNA configurations
2.3 基于TadA8e-dLbCas12a水稻基因组腺嘌呤碱基编辑的构建
本研究试图构建CRISPR/LbCas12a系统介导的腺嘌呤碱基编辑器,用于在水稻基因组中“AT”富集的区域引入A>G替换。本研究基于TadA8e建立了CRISPR/dLbCas12a介导的腺嘌呤碱基编辑器pUbi∶rBE58,并在水稻中检测其可行性和有效性。鉴于上述结果,本研究选择使用以TTTV为PAM的23 nt crRNA打靶3个水稻内源基因(OsCPK15,Bsrd1,OsJAZ4)(表2)。结果显示,在Bsrd1和 Os-JAZ4的靶位点没有检测到目的突变,仅有OsCPK15检测到目的突变,在获得的48个独立的转基因水稻品系中,鉴定出16个发生了A到G置换的植株(效率为33.33%),编辑框在TTTV PAM下游的+8-+14,活性窗口宽7 nt。其中,14个植株在PAM下游的第8位或第12位发生了单个靶碱基的置换,2个植株发生了多个靶碱基的置换事件,分别为PAM下游的第12、14位和PAM下游的第10、12、14位(图4)。这些数据表明,利用基于dLbCas12a的腺嘌呤碱基编辑器rBE58在水稻中实现定向碱基A向G的置换是可行的,但是存在位点依赖性。

图4 rBE58对水稻内源基因OsCPK15的靶位点腺嘌呤碱基编辑Fig.4 Targeted adenine base editing of endogenous gene OsCPK15 in rice using the rBE58 system

表2 实验中所用靶位点Table 2 Target sites used in this study
3 讨论
基于CRISPR/Cas12a建立的基因组编辑工具可实现靶位点的高效编辑,且以识别TTTV为PAM,弥补了CRISPR/Cas9系统的缺陷和不足,在动植物基因组编辑具有极大的潜力。据报道,在哺乳动物中,在20 nt crRNA的3′端加上U4AU4序列能改善其结合亲和力,促进细胞内crRNA和Cas12a蛋白的结合,可能有助于核糖核蛋白复合物形成的稳定,进而提高其编辑效率,但是却不影响脱靶效应[21]。然而在本研究中,CRISPR/LbCas12a和CRISPR/AsCas12a系统可以在水稻中实现基因编辑,但是crRNA的3′端富含“U”并没有明显改善其在水稻中的编辑活性,这可能是由于物种差异性造成的,同时crRNA的3′端富含“U”是否能提高编辑的特异性,即减少可能存在的脱靶效应需要进一步实验验证。
CRISPR/SpCas9及其突变体(SpCas9-NG和Sp-RY)或同源蛋白(ScCas9和SaCas9等)介导的腺嘌呤编辑器在水稻中已实现A·T到G·C,并将其识别的NGG PAM逐渐扩宽至NRN PAM,但是由于这些蛋白识别的PAM序列仍需要G或A的存在,使得Cas9介导的腺嘌呤碱基编辑器无法有效地实现富含AT的靶DNA(尤其是基因启动子)的腺嘌呤碱基编辑。本研究利用识别TTTV PAM序列的CRISPR/dLbCas12a系统建立了的水稻腺嘌呤编辑器rBE58,该编辑器的获得使得富含AT的靶DNA(尤其是基因启动子)的腺嘌呤碱基编辑成为现实,极大扩展了腺嘌呤碱基编辑技术在水稻中的应用和靶向范围。此外,rBE58与Cas9介导的腺嘌呤编辑器具有不同的碱基编辑活性窗口。本研究中,rBE58能够实现内源基因OsCPK15靶位点处PAM下游的第8、10、12和14位的靶碱基的定向替换,即其编辑活性窗口位于crRNA靶位点的中间位置(大约+10位置),这与已报道的Cas12a介导的动物细胞碱基编辑的编辑活性窗口一致[22],但与而传统的Cas9蛋白的活性窗口位于PAM的5′端存在差别。因此,rBE58丰富了ABE工具箱,为研究人员提供了更多的选择。然而,该编辑器在3个内源基因的检测中,仅有OsCPK15发生了A向G的置换,说明rBE58的编辑效率具有位点依赖性,这可能与染色质状态,DNA和/或组蛋白的修饰及dLbCas12a和腺嘌呤脱氨酶之间的相容性问题有关。
综上所述,本研究提出使用CRISPR/Cas12a系统在水稻中进行精准碱基编辑的思路,为将来开发更多与CRISPR/ Cas12a相关的工具用于基因功能研究和分子作物育种提供了良好的技术支撑。
4 结论
本研究以水稻为植物模型,通过筛选不同来源的Cas12a同源蛋白、crRNA长度、crRNA构型等因素,建立了高效的CRISPR/LbCas12a介导的水稻基因编辑系统,该系统识别TTTV为PAM,且在crRNA为23 nt时具有更高的编辑效率。基于dLbCas12a和腺嘌呤脱氨酶TadA8e成功建立的腺嘌呤碱基编辑器rBE58在水稻基因组中成功实现A>G碱基替换。
猜你喜欢
生物技术通报(2023年2期)2023-03-07 12:54:58
云南化工(2021年8期)2021-12-21 06:37:20
党的生活·党员电教与远程教育(2019年1期)2019-03-06 12:41:42
铁道通信信号(2018年7期)2018-08-29 01:17:08
电子设计工程(2015年4期)2015-02-27 12:04:14
中国药业(2014年21期)2014-05-26 08:56:45
天然产物研究与开发(2014年6期)2014-04-27 14:16:02
吉林大学学报(医学版)(2014年2期)2014-02-27 06:48:04
现代检验医学杂志(2014年6期)2014-02-02 03:01:54
食品科学(2013年15期)2013-03-11 18:25:42
