
Oncogenic driver mutations in non-small cell lung cancer: Past,present and future
2021-08-03 01:07:54MathieuChevallierMaximeBorgeaudAlfredoAddeoAlexFriedlaender
Mathieu Chevallier, Maxime Borgeaud, Alfredo Addeo, Alex Friedlaender
Mathieu Chevallier, Maxime Borgeaud, Alfredo Addeo, Alex Friedlaender, Department of Oncology, University Hospital Geneva, Geneva 1205, Switzerland
Alex Friedlaender, Department of Oncology, Clinique Générale Beaulieu, Geneva 1206,Switzerland
Abstract Lung cancer, of which non-small lung cancer is the most common subtype,represents the leading cause of cancer related-death worldwide. It is now recognized that a significant proportion of these patients present alterations in certain genes that drive oncogenesis. In recent years, more of these so-called oncogenic drivers have been identified, and a better understanding of their biology has allowed the development new targeted agents. This review aims to provide an update about the current landscape of driver mutation in non-smallcell lung cancer. Alterations in Kirsten rat sarcoma, epidermal growth factor receptor,MET, anaplastic lymphoma kinase, c-ROS oncogene 1, v-raf murine sarcoma viral oncogene homolog B, neurotrophic receptor tyrosine kinase, human epidermal growth factor 2, neuregulin-1 and rearranged during transfection are discussed, as well as agents targeting these alterations. Current standards of treatment as well as promising future strategies are presented. Currently, more than fifteen targeted agents are food and Drug administration-approved for seven oncogenic drivers in non-small-cell lung cancer, highlighting the importance of actively searching for these mutations. Continuous and future efforts made in defining the biology of each of these alterations will help to elucidate their respective resistance mechanisms, and to define the best treatment strategy and therapeutic sequence.
Key Words: Non-small cell lung cancer; Driver mutations; Tyrosine kinase inhibitors;Targeted agents; Oncogenes
INTRODUCTION
Lung cancer is the most common malignancy and the leading cause of cancer related deaths worldwide (18.4% of total cancer deaths), with non-small cell lung cancer(NSCLC) being the most common subtype, accounting for approximately 85% of all diagnosed cases[1]. The majority of NSCLC patients display advanced disease when diagnosed and thus have poor prognosis[2]. It is well established that acquired genetic alterations in certain driver genes result in tumor growth and invasiveness, and that patients harbouring certain mutations may benefit from targeted therapies[3](Figure 1).Indeed, a randomized clinical trial reported that advanced NSCLC patients harbouring activating mutations in theepidermal growth factor receptor(EGFR), one of the major oncogenic drivers of NSCLC, exhibited longer progression-free survival (PFS) when treated with a tyrosine kinase inhibitor (TKI), gefitinib, compared to those treated with standard platinum-based chemotherapy[4]. However, those who were treated with TKI drugs can acquire secondary resistance mutations, in which case a new treatment regimen is needed to maintain therapeutic effects[5,6]. In addition toEGFR, NSCLC patients carryinganaplastic lymphoma kinase(ALK) orc-ROS oncogene 1(ROS1)rearrangement were shown to respond well to a different TKI drug[7-9], crizotinib, while
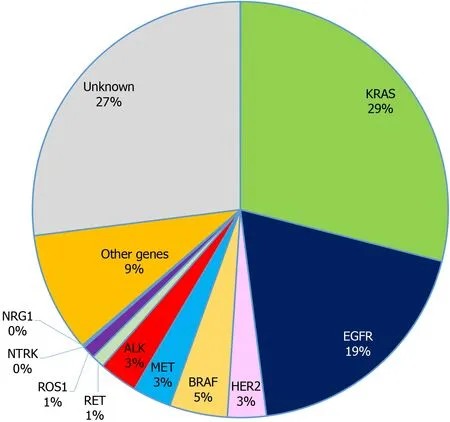
Figure 1 Incidence of oncogenic drivers in non-small cell lung cancer. KRAS: Kirsten rat sarcoma; EGFR: Epidermal growth factor receptor; ALK:Anaplastic lymphoma kinase; HER2: Human epidermal growth factor 2; ROS1: c-ROS oncogene 1; NTRK: Neurotrophic receptor tyrosine kinase; RET: Rearranged during transfection; NRG1: Neuregulin-1.
v-raf murine sarcoma viral oncogene homolog B(BRAF) mutated NSCLC patients can be treated with a combination of BRAF inhibitors, dabrafenib and trametinib[10]. These findings suggest that the identification of mutation profiles of NSCLC is critical in order to prescribe suitable TKI therapy as well as elucidate the molecular basis of drug resistance to provide timely treatment adjustment. Since 2018, the American Society of Clinical Oncology (ASCO) has recommended routine mutation testing for driver genes includingEGFR,ALK,ROS1andBRAFin clinical practice for patients with metastatic NSCLC. Although there are currently no targeted drugs forKirsten rat sarcoma(KRAS)orneuroblastoma rat sarcoma(NRAS) mutated NSCLCs[11,12], mutation testing for these genes has also been recommended due to their proven impact on clinical outcomes of NSCLC patients[13]. This review aims to provide an update about the impact and importance of detecting mutations in oncogenes in patients with advanced NSCLC.
KRAS
Therat sarcoma(RAS) genes (KRAS,NRAS,Harvey rat sarcoma viral oncogene homolog)represent the most frequent human oncogenes. Up to 30% of NSCLC harbor a mutation in theKRASoncogene, making KRAS the most commonly detected oncogenic driver in lung cancer[11]. The KRAS proteins belong to the small guanosine triphosphate (GTP)ase family, involved in intracellular signaling. In response to extracellular signaling, KRAS proteins switch between two states: The GTP-bound“on-state” and general dental practitioner-bound “off-states”. When “on”, KRAS activates downstream signaling pathways, mainly the mitogen activated protein kinase and extracellular signal regulated kinase (MAPK/ERK) and phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) C1 signaling pathways, ultimately promoting cellular division and proliferation.
Some KRAS mutations, such as those in exons 2 and 3, which prevent GTP hydrolysis and prevent switching KRAS signaling off, result in a constitutive activation of KRAS proteins. KRAS mutation is more frequent in adenocarcinoma, and can be detected by next generation sequencing (NGS)[14]. The most common mutations involve a substitution in the codon 12 or 13. The KRAS G12C mutation found in approximately 13% of NSCLC[15], is of particular interest, as it has become a therapeutic target. It is more frequently detected in smokers, while KRAS G12D is more common among non-smokers[11]
Currently, the standard of care for KRAS-mutated NSCLC follows that of nononcogene-driven NSCLC, consisting of immunotherapy with or without platinumbased chemotherapy.
Two Specific KRAS G12C TKIs have emerged, sotorasib (AMG510) and adagrasib(MRTX849). In the Codebreak100 phase I/II trial, 59 KRAS G12C mutated, previously treated advanced or metastatic NSCLC patients received sotorasib orally. The objective response rate (ORR) was 32.2%, with a median duration of response of 10.9 mo[16].
Sotorasib is currently being tested in the randomized phase III, Codebreak200 trial,vsdocetaxel in the second-line setting (NCT04303780). The primary endpoint is PFS,with overall survival (OS) as a secondary endpoint[17].
Adagrasib represents another specific KRAS G12C TKI. In the phase I/II KRYSTAL-1 trial[18], 79 patients with pre-treated NSCLC received adagrasib 600 mg twice daily.Among the 51 patients evaluable for response, an ORR of 45% was observed. The most frequent side effects included nausea, vomiting and diarrhea, mostly grade 1-2.
Others KRAS G12C inhibitors are currently being tested in phase I/II clinical trials:JNJ-74699157[19]and Gadolinia-Doped Ceria-6036[20]. As fewer than 50% of patients initially respond to sotorasib or adagrasib, we can assume that some patients present intrinsic resistance to KRAS G12C inhibition. This hypothesis is supported by preclinical evidence demonstrating resistant cell lines[21]. One explanation is that tumor cells may not exclusively rely on the RAS pathway for survival and proliferation[22]. As an example, RAS-independent activation of the PI3K/AKT/mTORC1 signaling pathway could be associated with resistance to KRAS inhibition[23]. Another mechanism of resistance could be the heterogeneous distribution of KRAS mutations in different tumor sites within the same patient[24]. Adaptive resistance also emerges under the selective pressure of KRAS TKIs. One mechanism of adaptive resistance could consist in the amplification of upstream drivers, such as receptor tyrosine kinases/ Src homology 2 domain-containing phosphatase 2 (RTKs/SHP2), that result from KRAS inhibition. Indeed, the diminution of ERK activity driven by KRAS G12C TKIs has been shown to suppress the ERK-mediated feedback inhibition of RTKs/SHP2, further activating N-Ras, H-Ras, and K-RasG12C, and ultimately restoring the activity of the MAPK/ERK signaling[21,25].
Although clinical data are scarce, it is likely that KRAS G12C inhibitors are not effective in the majority of the patients harboring KRAS G12C mutations. There has been a growing interest to combine the KRAS G12C inhibitors with targeted agents or immune checkpoint inhibitors (ICIs)[26]. Based on preclinical data discussed above,adagrasib is currently being tested in association with the SHP2 inhibitor TNO155 in early clinical phases[27]. Associations with ICIs also represent an interesting approach,as in some preclinical models, KRAS G12C positive tumor cell lines exhibit an immunosuppressive environment that is disrupted by KRAS inhibition[28,29].
Besides those targeting G12C, other KRAS inhibitors are in development, such as MRTX1133, a KRAS G12D inhibitor currently in investigational new drug enabling studies[30], or BI 1701963, a molecule targeting son of sevenless-1, an activator of KRAS,which could allow inhibition of the KRAS pathway regardless of the mutation[31].
Finally, other approaches targeting KRAS include an mRNA vaccine targeting KRAS mutant cells, a strategy that has already entered a phase I clinical trial[32], as preclinical data revealed an immune cell response in animal models[33].
EGFR
EGFR is one of the four members of the human epidermal growth factor (HER) family transmembrane receptors (HER1/EGFR, HER2, HER3, and HER4). Each HER receptor is an inactive monomer that dimerizes with a receptor of the same type or with another member of the HER family in response to ligand binding. The receptor activation triggers a complex downstream signaling network which leads cell replication[34]. The dysregulated receptor function or disruptions in any downstream EGFR processes may result in cell transformation and malignancy.
The prevalence of the mutation in the EGFR oncogenes is 50% among Asian patients with lung adenocarcinoma and 15% among Western patients[35]. Mutations leading to excessive EGFR activity are most common among non-smokers, young, female, Asian lung cancer patients[36]. Exon 19 deletions or L858R point mutations in exon 21 account for 90% percent of the activating mutations in the tyrosine kinase domain of EGFR,resulting in constitutional activation of EGFR without ligand-induced stimulation,thus promoting cell proliferation, survival, and dissemination[37,38].
EGFR mutations can be detected by immunohistochemistry (IHC) or NGS. TKIs are the standard front-line therapy for metastatic EGFR mutant NSCLC for a decade, with three generations of TKIs that demonstrated better outcomes and lower toxicity compared to standard chemotherapy[39], with a median PFS of 11.0 mo (gefitinib or erlotinib)vs5.6 mo (chemotherapy)[40]. To date, there are five United States Food and Drug Administration (FDA)-approved TKIs as the standard treatment for patients with activating EGFR mutations in NSCLC, including first-generation gefitinib and erlotinib, second-generation afatinib and dacomitinib, and third generation osimertinib[41]. However, it has been shown that resistance systematically develops to those treatments[42,43], mediated by mechanisms such as T790M secondary mutations,activation of other EGFR pathways, development of concurrent mutations or histological transformation. TheT790Mexon 20 mutation is rarely found in EGFR TKInaive disease but is the most frequent cause of resistance to first- and secondgeneration EGFR TKIs (50%-60% of cases)[44]possibly by the presence of a mutated clone before treatment, which would be free to grow into the dominant clone under selective TKI pressure. It is the indication for which osimertinib, an EGFR-TKI that selectively inhibits both EGFR-TKI-sensitizing and EGFR T790M resistance mutations,was first developed. It improves outcomes in second-line after disease progression withT790Mmutations[44,45], as well as in the front-line setting (PFS 18.9 movs10.2 mo;OS 38.6 movs31.8 mo) with an improved safety profile[43,46].
In order to improve outcomes of EGFR mutant NSCLC patients, combination therapy of EGFR TKIs with other agents have been tested, with interesting perspectives. A meta-analysis was conducted regarding the efficacy and safety of vascular EGFR inhibitors in combination with chemotherapy for patients with advanced NSCLC showing improved PFS, ORR and disease control rate, but without an impact on OS[47]. Results are pending from the Japanese phase II study comparing osimertinib alonevsosimertinib plus chemotherapy in second-line setting for patients with T790M mutation. Also, the phase III FLAURA2 study started recruiting in 2019,studying osimertinib with or without platinum-pemetrexed chemotherapy as first-line treatment inEGFRmutated advanced patientswith NSCLC (ClinicalTrials.gov Identifier: NCT04035486). Numerous studies are ongoing regarding the best sequence to use. At this time, the combination of chemotherapy and TKIs is not standard practice.
MET
The proto-oncogeneMETis located on chromosome 7q21-q31. It encodes for a transmembrane receptor (c-Met or MET) also known as hepatocyte growth factor receptor. This tyrosine kinase receptor activates downstream RAS/ERK/MAPK,PI3K/AKT, Wnt/β-catenin, and signal transducer and activator of transcription(STAT) signaling pathways, that can drive cell proliferation, survival, migration,invasion, angiogenesis, and transition from epithelial to mesenchymal[48]. MET dysregulation encompasses a heterogeneous array of alterations, with two main subgroups: MET amplifications and MET exon 14 mutations, leading to prolonged activation of the cellular MET receptor and downstream proliferation pathways[11].Regarding exon 14, aberrant splicing and skipping of exon in the messenger RNA transcript can result from somatic missense mutations, insertions, deletions, and concomitant insertions and deletions.
MET alterations are found in fewer than 5% of patients with NSCLC, mainly adenocarcinoma, often with concurrent mutations (i.e.,EGFR, ALK). Detection of MET amplification is assessed by fluorescencein situhybridization (FISH), and exon 14 skipping is most often completed through DNA or RNA NGS.
Due to the various diversity of MET dysregulation, there was a need to identify the oncogenic role of each type of MET alteration and defining the adequate targeted therapy. Over the last 20 years, several agents have been developed to target MET such as multikinase MET inhibitors (crizotinib, cabozantinib, MGCD265, AMG208,altiratinib, golvatinib), selective MET inhibitors (capmatinib, tepotinib, tivantinib) and monoclonal antibody (onartuzumab, emibetuzumab, ficlatuzumab, rilotumumab). A retrospective registry (IMMUNOTARGET) of advanced NSCLC patients who received immunotherapy showed that only 16% of them demonstrated a partial response, with a short median PFS of 3.4 mo[49]. Small cohorts showed that MET TKIs offer a promising treatment option in patients with exon 14 skipping with response rates from 25% to 68%, and a median duration of response of 9 to 16 mo. In the retrospective data analysis of a phase 1 PROFILE 1001 study, data supported that MET exon 14 skipping in NSCLC confers sensitivity to direct MET inhibitors with a median OS of 24.6 movs8.1 mo among patients receiving crizotinib compared with those who did not[50]. In patients with advanced or metastatic NSCLC with a confirmed MET exon 14 skipping mutation, the use of tepotinib, a highly selective oral MET inhibitor, was associated with a partial response in approximately half the patients (ORR 46%, with a median duration of response of 11.1 moin a recent phase 2 study[51]. In advanced MET exon 14 skipping NSCLC, capmatinib showed substantial antitumor activity particularly in previously untreated patients (ORR 68%), with a median duration of response of 12.6 mo[51]. Limited efficacy was observed in previously treated patients withMETamplification (ORR 7 to 12% of patients with capmatinib).
ALK
ALK is a transmembrane receptor tyrosine kinase that can activate multiple signaling cascades such as the PI3K-AKT, Crkl-C3G, MAP kinase kinase kinase 2/3-mitogenactivated protein kinase kinase (MEK)5-ERK5, Janus kinase (JAK)-STAT, and MAPK pathways. Its involvement is known in development, then subsequently silenced in adult tissues. However, severalALKgene alterations have been identified in tumors,including point mutations, deletions, and rearrangements leading to ALK reactivation.Various ALK-fusion proteins have been described that result from numerous chromosomal rearrangements, with formation of dimers by the amino-terminal portion of the ALK fusion proteins resulting in the activation of the ALK protein kinase domain that plays a key role in the tumorigenic process. The consequent ALK expression can activate multiple downstream known cancer signaling pathways[PI3K/AKT, JAK/STAT, and RAS/rapidly accelerated fibrosarcoma (RAF)/MEK/ERK][52].
ALK rearrangements are detected in approximately 5% of advanced NSCLC[8], 2 to 7% in all, up to 19% for stage IV. ALK alterations are mainly found in adenocarcinomas (97%), while squamous cell carcinomas comprise 3%[7]. ALK positivity is found in a fifth of never to light smokers with lung cancer. Methods of diagnosis can include FISH, IHC, or NGS.
The superiority of the TKI, crizotinib, over chemotherapy in first-line ALK+advanced NSCLC was proven in 2014, with an ORR of 74%vs45%, PFS 10.9 movs7 mo, and 1-year survival 84%vs79% (median OS not reached in the crizotinib group)[53]. However, resistance to treatment invariably develops, often with progression in the brain, motivating the development of new generations of TKI.Second- (alectinib, brigatinib, ceritinib) and third-generation (lorlatinib) TKIs have proven their efficacy, with the third-generation lorlatinib who led to a 72%improvement in PFS compared with crizotinib in first-line treatment[54]. After failure of second-generation ALK TKIs, concurrent administration of platinum/pemetrexedbased chemotherapy with an ALK TKI shows efficacy in a recent retrospective study[55](PFS 6.8 mo with the combinationvs3.2 mo for chemotherapy alone; suggesting a potential role for continued ALK inhibition. Some studies are currently underway,evaluating the addition of ceritinib to nivolumab (NCT02393625, completion June 2021) or ceritinib with trametinib (NCT03087448, completion June 2022).
Recently, a next-generation ALK inhibitor, ensartinib, demonstrated promising efficacy in the first-line treatment for advanced disease in a preplanned interim analysis (phase III eXalt3 study), with a median PFS of 25.8 mo with ensartinib in the intent-to-treat populationvs12.7 mo with crizotinib) (NCT02767804, study completion March 2021). Another new ALK inhibitor, TQ-B3139, is being testedvscrizotinib in the first line setting (NCT04009317, completion April 2022).
ROS1
ROS1 encodes a tyrosine kinase receptor, belonging to the insulin receptor family, and structurally related to the ALK protein. Its natural ligand remains undefined.
ROS1 rearrangement was described in glioblastoma, before being recognized as a potential oncogenic driver in NSCLC[56]. Various ROS1 alterations have been described in cancers: overexpression, splice variants (usually leading to a truncated protein lacking the intracellular domain), amplification, mutations and finally fusions with another partner gene. In contrast to fusions, the pathogenic significance of other alterations is undetermined[57].
At the moment, ROS1 fusions have identified with 55 different partners genes in different cancer types, including more than 20 fusion partners in NSCLC. The CD74-ROS1, EZR-ROS1, SDC4-ROS1 and SLC34A2-ROS1 fusion represent the most common rearrangements[58]. All resultant fusion proteins retain an intact ROS1 intracellular kinase domain which, as a result of the rearrangement, becomes constitutively activated. The activated kinase triggers intracellular signaling pathways, such as the RAS-RAF-MEK-ERK, PI3K-AKT-mTOR and JAK-STAT3 pathways, ultimately leading to cellular survival and division.
As with other common driver mutations, patients harboring a ROS1 rearrangement tend to be younger and more frequently non-smokers and Asian[56]. ROS1 is almost always found in adenocarcinoma subtypes, where it represents 1%-2% of the cases[59].ROS1 alterations rarely occur with other driver mutations[60].
ROS1 rearrangement can be detected by FISH or NGS[14]. IHC is sensitive and can be used as a screening test. However, specificity remains poor and a positive result must be confirmed by FISH assays or NGS. A treatment decision should not be based on IHC results alone.
ROS1 rearranged NSCLC seem to have better response to chemotherapy, especially pemetrexed-based, when compared to both tumors with other driver mutations and wild-type tumors[61]. On the other hand, some data, although scarce, show modest response to ICIs[62].
Several tyrosine kinase inhibitors have shown clinical activity in ROS1-positive NSCLC. The phase I PROFILE 1001 trial of crizotinib included 50 patients with ROS1-fusion NSCLC, among which an overall response rate of 72%, and a median PFS of 19.2 mo were reported[63]. Importantly, more than 80% of patients had received at least one previous line of treatment. An updated analysis reported an impressive median OS of 51 mo[64]. Phases 2 trials in Europe and East Asia have consistently reported similar ORRs of 63%-69%[65,66]. In these studies, crizotinib demonstrated a favorable safety profile. However, crizotinib has limited intracranial activity. The central nervous system represents a common site of progression in ROS1-positive NSCLC treated with crizotinib[67]. Crizotinib should be avoided as a first-line agent in case of untreated brain metastasis.
After progression on crizotinib, lorlatinib represents an option. It was evaluated in 69 patients with ROS1-positive NSCLC[68]. In 40 patients that had received prior crizotinib, the ORR was 35%, with a median PFS of 8.5 mo. In 21 crizotinib-naive patients in the same study, lorlatinib demonstrated an ORR of 62%, with a median duration of response comparable to crizotinib of 25.3 mo and a median PFS of 19.3 mo.Lorlatinib also showed intracranial activity, with intracranial response rates of 50%and 64% in crizotinib pretreated and crizotinib-naïve patients respectively.
Ceritinib is a selective ALK inhibitor that also exhibits activity against ROS1 kinase.In a phase phase II trial involving 32 Asian patients with ROS1-rearranged NSCLC the ORR was 67%[69]. Interestingly, two patients that had received prior crizotinib experienced no response.
Entrectinib represents another tyrosine kinase inhibitor with activity against the ROS1 kinase. In a phase II study among 53 patients with ROS1-positive NSCLC, the majority of which had received previous platinum-based chemotherapy, the ORR reached 77%[70]. Responses seem durable, with median duration of response of 24.6 mo.Intracranial activity was also observed, with an intracranial ORR of 55%, with a median duration of response for central nervous system lesions of 12.9 mo.
Repotrectinib represents a next generation TKI targeting ROS1, ALK and tropomyosin receptor kinase (TRK). Its potency for ROS1 exceeds that of crizotinib by more than 90-fold. Reprotrectinib also shown early signs of efficacy against ROS1 resistance mutations[70,71].
Resistance to TKIs ultimately arise in nearly all patients. Intrinsic resistance to ROS1 inhibition implies solvent-front or gate-keeper point mutations in the ROS1 kinase domain, the most common being the ROS1 G2032R, that precludes binding to crizotinib[72]. To date, more than 20 mutations conferring resistance to various TKI have described[57]. Moreover, resistance to ROS1 inhibition may probably arise from the activation of parallel signaling pathways such as KRAS, BRAF or MET. Emergence of BRAF and KRAS activating mutations and MET-amplification in ROS1 positive NSCLC treated with crizotinib or lorlatinib have been described[73,74]. Upon progression, the role of sequencing ROS1 TKIs remains unclear.
BRAF
BRAF is an intracellular protein kinase that plays a relevant role in the MAPK/ERK pathway, including numerous proteins with kinase domains (RAF, MEK, ERK) that carry signal transduction from membrane receptors to DNA in the nucleus of the cell[75]. BRAF is an oncogene located on chromosome 7 involved in several cell functions, including growth, proliferation, survival and differentiation.
BRAF mutations are found in about about 5.5% of cancers, with almost 200 BRAF mutants and many RAF translocations identified in human cancers[76]. Those alterations generate structural modifications of the protein that are responsible for permanent activation of MAPK pathway and resistance to inhibitory feedback signals.
The frequency of BRAF mutations is about 5 to 8% in lung adenocarcinomas[10], with higher incidence in melanoma (50%), thyroid carcinoma (30% to 70%) and colorectal cancer (5% to 20%). Its presence predicts poor outcome for the latter three. While the V600E activating mutation is the most common BRAF variant found in solid tumors(90%), it only accounts for half of BRAF mutations in NSCLC. Non-V600E variants are more common in males, and all BRAF variant are more common among smokers. It is important to note that clinical characteristics of patients with NSCLC harboring BRAF mutation are difficult to clearly identify due to small numbers of patients in trials.
BRAF mutations can be detected by using immunohistochemistry for V600E exclusively or DNA sequencing on the tumor tissue for the two types.
The efficacy of ICIs is uncertain, and is based on conflicting retrospective data and case series, thus not routinely recommended. Therapies targeting BRAF mutations were developed for melanoma with great success, then used for NSCLC. For lung cancer, numerous small studies tested RAF inhibitors (vemurafenib, dabrafenib) alone or in combination with MEK inhibitors (trametinib, cobimetinib). Best responses were observed when treatment was combined, especially in V600E patients in first-line of dabrafenib-trametinib with a mOS of 24.6 mo[77]. Front-line use of double BRAF/MEK inhibition if V600E mutation is found in advanced NSCLC is now suggested in current guidelines. Other agents such as multikinase inhibitors sorafenib and dasatinib showed responses in case reports[78,79].
Combining MEK and BRAF inhibitors has proven to be more effective than singleagents for the treatment of BRAF-mutant advanced tumors, but does not prevent the emergence of resistance[79]. Indeed, studies on melanoma describe intrinsic adaptive(other pathways alteration) or acquired (de novo) resistance in BRAF mutated cancers treated with targeted inhibitors. Thus, further research is warranted to establish clear therapeutic algorithms.
NEUROTROPHIC RECEPTOR TYROSINE KINASE
The threeneurotrophic receptor tyrosine kinase(NTRK) genes (NTRK1,NTRK2andNTRK3) encode tyrosine kinase receptors for neutrophins, involved in neuronal development, survival and proliferation[80,81].
Oncogenic fusions involving the NTRK genes (e.g.,apposition of the 3′ region of the NTRK gene with the 5′ sequence of a fusion partner gene) occur across many cancer types[82]. More than 25 gene partners have been described[83]. Yet, all these fusions result in a constitutively activated and overexpressed TRK kinase, that activates downstream pathways involved in cellular proliferation, such as the MAPK and PI3K/AKT[84].
NTRK fusions are very frequent in a few rare cancer types: Secretory carcinoma,mammary analogue secretory carcinoma, infantile fibrosarcoma and cellular mesoblastic nephromas, in which they are detected in more than of 90% of patients[83].On the other hand, the NTRK fusion occurs at a very low frequency in various common cancers types, such as colorectal, breast, thyroid and lung cancers. In the latter, it represents under 1% of cases[85,86].
NTRK fusion can be detected either by IHC, FISH or DNA-and RNA-based NGS.Pan-TRK IHC has been shown to have high specificity and sensitivity in the detection of fusion protein expression. RNA-based NGS is preferred to DNA-only based technique.
The European Society for Medical Oncology (ESMO) guidelines suggest using FISH testing (or real-time reverse transcription polymerase chain reaction) in tumors with highly prevalent NTRK fusions, as it probably represents the most cost-effective strategy. In situations where such alterations are uncommon, NGS technique should be used upfront if available or as a confirmatory test, after a positive IHC “screening test”. Using NGS upfront has the advantage to allow the detection of other potentially targetable molecular alterations.
Larotrectinib is an oral TRK inhibitor that has shown durable activity in NTRK positive advanced tumors. In an analysis of three phase I/II trials, the ORR among patient with TRK-fusion positive cancer was 79%, with 80% of responses ongoing at 12 mo[15]. Among 12 patients with lung cancer, a similar response rate was reported (75%).The most frequent grade 3-4 adverse events related to larotrectinib were increased alanine aminotransferase, anemia, and decreased neutrophil count. Grade 3-4 adverse events occurred in 46% of patients.
Entrectinib represents another TRK inhibitor, also active for ROS1 and ALK, and specifically designed to cross the blood-brain barrier[87]. Entrectinib demonstrated an ORR of 57% in small non-randomized studies, among 54 patient with TRK-positive advanced tumors, including 10 patients with NSCLC (ORR 70% in the latter)[87]. The median overall duration of response was 10 mo. Of note, an objective intracranial response rate of 54.5% was reported in a cohort of 12 patients with NTRK fusionpositive tumors with brain metastasis[88]. The most common adverse events reported with entrectinib (> 20% of patients) were fatigue, gastro-intestinal disorders, weight gain and cognitive impairment. While the most threatening serious adverse events consisted of congestive heart failure, QT prolongation, central nervous system effects,hepatotoxicity, and vision disorders.
Based on these results, both larotrectinib and entrectinib have been granted accelerated approval by the FDA for metastatic or unresectable NTRK-fusion positive solid tumors, that have progressed following treatment or have no satisfactory standard therapy[89,90].
Despite a prolonged response, resistance to larotrectinib and entrectinib is expected to emerge in most patients[83],viadifferent mechanisms: Solvent-front mutations and xDFG substitution[91]. Larotrectinib (LOXO)-195 and repotrectinib represent secondgeneration TRK-inhibitors, currently under development capable to overcome resistance to first-generation of TRK-inhibitors. LOXO-195 is a highly selective inhibitor of all 3 TRK kinases. LOXO-195 or selitrectinib was evaluated in a phase I trial and a FDA expanded access single patient protocol, including 31 patients in total,all of whom had received prior treatment with a TRK inhibitor[92]. Of note, 7 patients were pediatric. The most frequent treatment-emergent adverse events were dizziness(65%), ataxia (60%), nausea (50%), vomiting (40%), anemia (30%) and gait disturbance(30%), with 5 double-lumen endobronchial tube (4 ataxia/dizziness) in total. Among the 29 patients evaluable for efficacy, an ORR of 34% was reported.
Repotrectinib[91]is another second-generation NTRK inhibitor, designed to overcome acquired resistance mutations to larotrectinib/entrectinib[93,94]Case reports showed partial response in patient presenting acquired resistance to larotrectinib or entrectinib[87]. Repotrectinib is currently being tested in phase I/II clinical trial(NCT03093116)[95].
HER2
TheHER2/neu gene is located on the chromosome 17q12, and encodes the HER2 protein, which belongs to the epidermal growth factor receptor (ERBB) family of tyrosine kinase receptors. Other members of the ERBB family include EGFR (ERBB-1),HER3 (ERBB3), and HER-4 (ERBB4). The ERBB receptors are transmembrane proteins,composed of an extracellular ligand binding domain, an α-helical trans membrane segment and an intracellular tyrosine kinase domain. Ligand binding induces receptor dimerization, and auto-phosphorylation and activation of the intracellular kinase domain[96]. Of note, no natural ligand of HER2 has been identified. Nevertheless, HER2 can undergo dimerization with other ERBB receptors[97], and represents their preferred dimerization partner. The activated kinase domain that results then triggers downstream signaling pathways, such as MAPK, PI3K/AKT, protein kinase C and STAT, promoting cellular proliferation and inhibiting apoptosis[98]. Genetic alterations in HER2 can result in constitutive dimerization and activation of downstream signaling pathways, finally leading to uncontrolled cellular proliferation.
HER2 mutations is found in approximately 1%-3% of NSCLC, primarily in adenocarcinoma, in non-smokers and women[99]. Mutations usually consist of in frame insertions or point mutation in exon 20. On the contrary, HER2 amplification in NSCLC is not associated with benefit of anti-HER2 therapy[100].
Retrospective data indicate that HER2 targeted therapy present some degree of activity in HER2-mutated NSCLC. In a European cohort of 57 pre-treated HER2 mutated NSCLC patients receiving trastuzumab-based regimen, an ORR of 50% was reported, with a PFS of 5.1 mo[101]. In a prospective phase II trial, the antibody-drug conjugate ado-trastuzumab emtansine demonstrated an ORR of 44% in NSCLC patients harboring HER2 mutations, with a median PFS of 5 mo[102]. The most common treatment-related adverse events included grade 1 or 2 infusion reactions,thrombocytopenia, and elevated aspartate aminotransferase or alanine aminotransferase. No grade 4 or 5 adverse events were reported. More recently, the DESTINY-Lung01 trial evaluating the safety and efficacy of trastuzumab-deruxtecan,another antibody-drug conjugate targeting HER2, were presented at the ASCO meeting[103]. The DESTINY-Lung01 trial includes 2 cohorts: HER2 amplified (based on IHC) and HER2 mutated tumors[104]. Interim results concerning only the HER2 mutated population were reported. Of the 42 patients who received trastuzumab-deruxtecan,with a median of 2 prior treatment lines, the confirmed ORR was 61.9%, with a median duration of response not reached at data cut-off, and a median PFS estimation of 14 mo. However, toxicity was not negligible, as 64.3% of patients presented grade 3 or more adverse events (52.4 % drug-related). Of note, 11.9% of developed drug-related interstitial lung disease, all grade 2. Treatment related adverse events led to treatment interruption, dose reduction or treatment cessation in 59.5%, 38.1% and 23.8% of patients respectively. The randomized phase II DESTINY-Lung02 trial (NCT04644237)will compare a lower dose regimen of 5.4 mg/kg every 3-wk (Q3W) to the 6.4 mg/kg Q3W regimen used in DESTINY-Lung01 trial[105].
NEUREGULIN-1
Theneuregulin-1(NRG1) gene is located on the long arm of chromosome 10 (10q23.1 region) and encodes a growth factor belonging to the complex family of proteins called heregulins, structurally related to the stimulation of ERBB receptors tyrosine kinase activity and EGF signals[106]. NRG1-receptor binding activates the ERBB2-ERBB3 heterocomplex and controls proliferation, differentiation, and survival in both normal and tumor cells through the predominant signaling cascades PI3K-AKT and MAP kinase[107].
NRG1 oncogenic gene rearrangements or fusions are rare, with an incidence of 0.2%on a wide retrospective molecular profiling that tested over 21’850 solid tumors[108].They have been identified across a wide range of tumors including NSCLC (especially mucinous adenocarcinoma subtype), gallbladder cancer, pancreatic cancer, renal cell carcinoma, ovarian cancer and hepatic cholangiocarcinoma[109].
Detection of NRG1 gene fusions in solid tumors are made through RNA NGS[110].NRG1aberrations appear to be mutually exclusive with oncogenic alterationsin EGFR,KRAS, ALK, ROS1, andrearranged during transfection(RET).
NRG1was first described in NSCC in 2014[110]. Invasive mucinous adenocarcinoma(IMA), representing approximately 5% of lung adenocarcinomas, are known to be more aggressive than more common types, such as acinar or papillary adenocarcinoma.KRASmutations had been the only oncogenic driver commonly detected in IMAs (in 50%-80% of cases), butCD74-NRG1fusions are now detected in 14.7% ofKRASnegative IMAs. NRG1 positivity confers worse outcomes in lung adenocarcinoma in retrospective data[111]. Given the potential therapeutic implications of this genetic alteration, the interest in evaluating the prevalence of NRG1 fusions has increased over the last five years.
The use of afatinib (tyrosine kinase inhibitor, targeting ERBB) showed interesting results in case reports for the treatment ofNRG1fusion-positive in cancers of lung (SDC4-, SLC3A2-andCD74-NRG1gene fusion) and hepatocellular (ATP1B1-NRG1gene fusion) origin[112]. Further studies are ongoing.
RET
TheRETgene, located on the chromosome 10 (10q11.2), encodes a tyrosine kinase receptor located to the cell surface. Its intracellular kinase domain shares 37%homology with the ALK kinase domain[113]. RET is a receptor for Glial Cell Line-Derived neurotrophic factor (GDNF), a family extracellular signaling molecules notably involved in neuronal development[114]. The binding of GDNF to a co-receptor GDRalpha and then to RET, leads to a RET-dimerization and further autophosphorylation of the kinase domains. This activates the intracellular signal transduction process, notably the RAS, MAPK/ERK, PI3K/AKT and JAK/STAT pathways[113]. RET is normally involved in enteric nervous system and urogenital tract development. RET loss of function is associated with Hirschprung disease[115], and activating mutations to MEN-2 syndrome[116]. Rearrangements of theRETgene (e.g.,the apposition of the Cterminal region of RET protein with the N-terminal region of another protein) can induce a constitutive activation of the RET kinase.
Rearrangements in RET with various partners have been described, the most common in NSCLC being KIF5B and CCDC6[117]. They have been identified in 1%-2%of lung adenocarcinomas, more often in never smoker and younger patients[113]. IHC is probably unreliable for the detection of RET-rearrangements, and FISH or NGS are preferred[113].
The phase I/II LIBRETTO 001 trial[118]evaluated selpercatinib in NSCLC patients. It showed an ORR of 64% in 105 patients previously treated with platinum-based chemotherapy and 85% in previously untreated patient (39 patients). Median duration of response was 17.5 mo in previously treated patients. Interestingly, among the 11 patients with central nervous system disease, 91% had an intracranial response.
Following these results, selpercatinib was granted accelerated approval by the FDA[119]. Selpercatinib is currently being evaluated in phase III,vsplatinum-based chemotherapy +/- pembrolizumab, in the LIBRETTO-431 trial[120].
Pralsetinib (BLU-667), another selective RET inhibitor has also been granted accelerated approval from the FDA based on the results of the ARROW-study(NCT03037385), a basket trial in which patients with RET-fusions positive cancer received pralsetinib 400mg orally once daily. Among 89 patients with RET-fusion positive NSCLC pretreated with platinum-based chemotherapy, ORR was 57%, with 80% of ongoing responses at 6 mo[121]. Among 27 patients with previously untreated NSCLC, the ORR was 70% with 58% of response ongoing at 6 mo. Praseltinib is currently being compared to the standard treatment approach of platinum-based chemotherapy ± pembrolizumab in RET-fusion positive NSCLC in the AcceleRET trial(NCT04222972)[122].
Other non-RET-selective TKIs have shown some activity in RET-fusion positive NSCLC: Cabozantinib for example displayed an ORR of 28% in a single-center, phase II trial, of patients with RET-rearranged NSCLC, of whom 75% had received prior chemotherapy[123]. In an international registry of patients with RET-rearranged NSCLCs, cabozantinib, vandetanib, and sunitinib had rather limited activity, with overall response rates of 37%, 18%, and 22%, respectively[124].
Different mechanisms of RET-inhibition resistance have been described including the emergence of solvent front mutation in theRET-gene (e.g.in the RET G810 residue,in the kinase solvent front)[125]as well as acquired MET or KRAS amplifications[126].
CONCLUSION
Driver mutations have significantly altered the diagnostic work-up and reshaped the oncology treatment paradigm. Recently, several new driver mutations have been identified in metastatic NSCLC, with some leading to therapeutic success and others,failure. We have summarized the current landscape of actionable oncogenic alterations and their therapies in Table 1. A better understanding of the biology of various subtypes of each driver mutation will help not only to match the optimal treatment to each patient, but also elucidate their respective resistance mechanisms, allowing for greater precision medicine. Many trials are ongoing, some of them through serial tumor or plasma biopsies and multiplex molecular testing. The optimal targeted therapy sequence for each driver mutation is yet to be fully determined. Furthermore,it is unclear whether a multi-kinase inhibitor or highly selective therapy is the best choice for some alterations, though the latter tend to have more favourable toxicity profiles.

Table 1 Selected oncogenic drivers and their treatments in non-small cell lung cancer
Given the portfolio of possible mutations and targeted therapies to offer, multiplex NGS testing should be standard practice. Barring a therapeutic emergency, no patient should be started on systemic therapy before a comprehensive molecular analysis has been completed. In the twentieth century, every gene matters, and it would be unethical to deny patients access to proven targeted therapies given the efficacy and favourable toxicity profile of such drugs.
 World Journal of Clinical Oncology2021年4期
World Journal of Clinical Oncology2021年4期
- World Journal of Clinical Oncology的其它文章
- Tongue swelling as a manifestation of tongue metastasis from pulmonary sarcomatoid carcinoma: A case report
- Lenvatinib-induced multiorgan adverse events in Hurthle cell thyroid cancer: A case report
- Hepatocellular carcinoma with biliary and neuroendocrine differentiation: A case report
- Positron emission tomography complete metabolic response as a favorable prog-nostic predictor in esophageal cancer following neoadjuvant chemotherapy with docetaxel/cis-platin/5-fluorouracil
- Cytotoxic CD8+ T cells and tissue resident memory cells in colorectal cancer based on microsatellite instability and BRAF status
- GOECP/SEOR radiotherapy guidelines for thymic epithelial tumours