
BRCA mutations and gastrointestinal cancers: When to expect the unexpected?
2021-08-02 03:13:18ElenaMaccaroniRiccardoGiampieriEdoardoLenciLauraScortichiniFrancescaBianchiLauraBelvederesiCristianaBrugiatiSilviaPagliarettaElisaAmbrosiniRossanaBerardi
Elena Maccaroni, Riccardo Giampieri, Edoardo Lenci, Laura Scortichini, Francesca Bianchi, Laura Belvederesi,Cristiana Brugiati, Silvia Pagliaretta, Elisa Ambrosini, Rossana Berardi
Elena Maccaroni, Department of Medical Oncology, Azienda Ospedaliero-Universitaria Ospedali Riuniti di Ancona, Ancona 60126, Italy
Riccardo Giampieri, Edoardo Lenci, Laura Scortichini, Rossana Berardi, Department of Medical Oncology, Università Politecnica delle Marche, Ancona 60126, Italy
Francesca Bianchi, Laura Belvederesi, Cristiana Brugiati, Silvia Pagliaretta, Elisa Ambrosini,Dipartimento di Scienze Cliniche e Molecolari, Università Politecnica delle Marche, Ancona 60126, Italy
Abstract BRCA1/2 pathogenic variants are widely known as major risk factors mainly for breast and ovarian cancer, while their role in gastrointestinal (GI) malignancies such as colorectal cancer (CRC), gastric cancer and oesophageal cancer (OeC) is still not well established. The main objective of this review is to summarise the available evidence on this matter. The studies included in the review were selected from PubMed/GoogleScholar/ScienceDirect databases to identify published articles where BRCA1/2 pathogenic variants were assessed either as a risk factor or a prognostic/predictive factor in these malignancies. Our review suggests that BRCA1/2 might have a role as a risk factor for colorectal, gastric and OeC, albeit with differences among these diseases: In particular BRCA1 seems to be much more frequently mutated in CRC whereas BRCA2 appears to be much more closely associated with gastric and OeC. Early-onset cancer seems to be also associated with BRCA1/2 mutations and a few studies suggest a positive prognostic role of these mutations. The assessment of a potentially predictive role of these mutations is hampered by the fact that most patients with these diseases have been treated with platinum compounds, where it is expected that a higher probability of response should be seen. A few clinical trials focused on poly (ADPribose) polymerase inhibitors use in GI cancers are currently ongoing.
Key Words: BRCA; Colorectal cancer; Gastric cancer; Esophageal cancer; Prognosis; Poly(ADP-ribose) polymerase inhibitors
INTRODUCTION
BRCA mutations are defined as pathogenic variants in either theBRCA1orBRCA2gene: These two tumour suppressor genes are involved in different crucial pathways including DNA repair, cell proliferation control and apoptosis. In particular,BRCA1andBRCA2genes are mainly involved in the homologous recombination (HR) process,responsible for maintenance of genome integrity through an error-free repair pathway for DNA double-strand breaks in response to DNA damage[1,2]. Loss-of-function mutations in BRCA1/2 may lead to accumulation of DNA double-strand breaks and result in genomic instability and tumour development.
BRCA1 and BRCA2-associated hereditary breast and ovarian cancer syndrome is characterised by an increased risk of breast cancer and ovarian cancer (including fallopian tube and primary peritoneal tumours), up to 84% for breast cancer and 40%for ovarian cancer[3-5]. Moreover, higher risk of male breast cancer has been reported especially in BRCA2 carriers[6-9].
In BRCA2 pathogenic variant carriers, familial cancer types different from breast and ovarian, such as prostate, pancreatic cancer, and melanoma, have also been described[10-14].
In addition to that, case-control and family-based studies seem to suggest that BRCA1/2 pathogenic germline carriers might be at increased risk of other malignancies, although this seems less likely[6,15,16]. It is unclear whether BRCA1 and BRCA2 mutations might determine increased risk of tumour types that are already quite common in the general population, such as gastrointestinal (GI) cancers: This might be due to the fact that most data on the subject is derived from cross-sectional studies of family histories of women with BRCA pathogenic mutations.
These studies might be susceptible to selection bias; furthermore, misclassification can be present, due to the fact that GI cancer diagnoses were based only on information provided by a family member and this approach may not always be reliable.
Moreover, prospective evidence estimating GI cancer incidence in BRCA1/2 pathogenic variants carriers are still rare[17]. Predicting GI cancer incidence rates among BRCA carriers might convey meaningful implications both for genetic counsellors and tested individuals, as it would aid in the development of appropriate screening policies and risk reduction procedures. Indeed, at the moment, specific guidelines or recommendations regarding the need for gastric and bowel screening procedures for carriers of BRCA1/2 mutations are lacking.
Furthermore, BRCA1 and BRCA2 pathogenic variants have an emerging role for novel target therapies. BRCA1/2 have essential functions in the HR DNA repair pathway, and genomic alterations in BRCA genes lead to impaired DNA repair, called homologous recombination deficiency (HRD). Platinum compounds [such as oxaliplatin, widely used in colorectal and gastric cancer (GC) treatment] and poly (ADPribose) polymerase-inhibitors (PARPi) are currently the two main classes of drugs active against cancer cells harbouring HRD alterations. In particular, PARPi exposure promotes synthetic lethality in the setting of defective DNA repair pathway due to irreversible DNA damage, and resulted extremely effective in BRCA-mutant ovarian cancer (OvC): In platinum-sensitive OvC patients PARPi maintenance therapy now represents a mainstay of treatment (Figure 1). Currently PARPi use has also been approved in BRCA-mutant breast, pancreatic and prostate cancer patients[18-21].

Figure 1 BRCA, PARP-inhibitors and synthetic lethality in gastrointestinal cancers.
In BRCA-mutant GI cancer, the role of HRD alterations is still widely unknown and only few data about their clinical impact are available.
The aim of this review is to highlight a possible association between BRCA pathogenic variants and GI cancer risk and also to investigate the role of BRCA mutations as a prognostic and/or predictive factor in this setting of patients. As the role of BRCA mutations (particularly BRCA2) has already been clarified in pancreatic cancer patients and in such cases PARPi[20] are already used in everyday clinical practice, we decided to focus only on gastric, oesophageal cancer (OeC) and colorectal cancer (CRC).
LITERATURE SEARCH
The studies included in the present review were selected from PubMed/Google-Scholar/ScienceDirect databases. We used the terms “Gastric Cancer”, “Colon Cancer”, “Rectal Cancer”, “Colorectal Cancer”, “Esophageal Cancer”, “Oesophageal Cancer” for tumour location and also the terms “BRCA1”, “BRCA2”, “BRCA” to select papers focused on BRCA mutations. “Germline” was added as a search term to identify published papers concerning familial BRCA mutations (as opposed to papers where somatic BRCA mutations or all kinds of mutations regardless of their type were included). No limitation as to the year of the published paper was used. Only published papers were included in the analysis (meeting abstracts were excluded).Duplicates were eliminated as well as articles not written in English. The search for articles was conducted independently by two of the co-authors (Maccaroni E and Giampieri R).
CRC
CRC in BRCA carriers: Risk and clinico-pathological features
CRC is the third most common malignancy in both sexes: It accounts for 10% of new cancer diagnoses and it represents the second estimated leading cause of cancerrelated death in 2020 worldwide[22].
One out of 20 CRC patients has a hereditary predisposition. Lynch syndrome (LS,hereditary non-polyposis CRC) is the most common hereditary disorder, justifying 3%of all cases; LS is an autosomal dominant syndrome that causes alterations in the DNA repairing system known as mismatch repair (MMR) proteins. These proteins are able to restore insertions, deletions, as well as A-G and T-C mismatches[23]. Lack of function variants in one of the four genes (hMLH1, hMSH2, hMSH6, PMS2) that encodes the MMR proteins determines, as a final effect, an increase in replication errors in specific areas of the genome where single or double-nucleotide repeats are present, known as microsatellite regions. An increase or decrease in microsatellite length results in what is known as microsatellite instability (MSI) and increases DNA susceptibility to further mutation in oncogenes and tumour suppressor genes. Twelve to fifteen percent of all CRCs harbour a deficient MMR system and the percentage is lower (5%) if we only consider patients with metastatic involvement (mCRCs)[24].Among these cases, 2/3 are due to sporadic mutations, while only 1/3 of cases are due to germline loss-of-function mutations. Because of this fact, patients with MSI-high status CRC diagnosis should be advised genetic counselling and testing so as not to overlook further diagnosis of LS. Other clinical factors that might lead to suspected LS are Amsterdam I/II criteria and revised Bethesda guidelines[25,26]: Although these criteria might suggest the need for pedigree analysis, it must be considered that genomic profiling remains the gold standard for diagnosis of LS either in patients who fit these criteria or those screened because of a MSI-high CRC tumour.
While the risk of developing CRC in LS is well known, the same does not apply to BRCA1/2 mutation carriers; several efforts have been made to evaluate the lifetime susceptibility of BRCA1/2 mutation carriers to develop CRC and have yielded conflicting results.
Mauriet al[27] suggested that BRCA1/2 mutations might determine an increase in CRC diagnoses, particularly in young patients.
Another study of Kimet al[28] suggested that even first and second-degree relatives of high-risk BRCA mutated (BRCAm) breast cancer patients are at increased risk of non-breast/ovarian cancer.
Phelanet al[17] conducted a multicentre prospective study involving a cohort of 7105 female patients with the aim of assessing the CRC incidence in women carrying pathogenic BRCA1/2 mutations. Enrolment was conducted in fifty centres among five countries in Canada, United States and Europe, from 1992 to 2010; the median followup was 5.5 years.
When newly diagnosed CRC were matched with population-specific incidence rates, no statistically significant differences were found: Authors reported 21 new CRC diagnoses between mutation carriers, while 23.6 cases were expected. Among them, 16 cases occurred in BRCA1m (vs17.4 expected), and 5 cases in BRCA2m (vs6.1 expected). Although this study failed to show a higher prevalence in all mutated patients, subgroup analysis of BRCA1 carriers showed that early-onset (30-49 years)CRC diagnosis was four times greater than controls. The large population of patients enrolled and the prospective design of this study support these findings; however, the lack of male patients narrows these results to only the female population. Moreover,the relatively small number of incident cases and the ability to retrieve only 2/3 of the pathological reports, may represent another limitation of the study.
Similarly, Ohet al[29] presented a meta-analysis of 14 out of 18 studies initially selected, as to estimate the CRC risk in BRCAm carriers. Using a random-effects model, the authors observed a statistically significant increase in the odds of CRC in BRCAm carriers [odd ratio (OR) = 1.24, 95%CI: 1.02 to 1.51,P= 0.03]. When the authors focused on specific BRCA1 or BRCA2 carriers, they highlighted that BRCA1 carriers were at a higher risk of developing CRC. On the other hand, they did not observe a higher risk of early onset CRC as previously stated by Phelanet al[17].
In contrast, in their systematic review and meta-analysis Cullinaneet al[30] did not observe any statistically significant difference in terms of CRC risk among BRCAm carriers, regardless of the gene involved. Neither did adjustment for age modify these findings, nor when the analysis was conducted only in the Ashkenazi Jewish heritage population.
It is well established that BRCA1/2 mutations incidence is broader in some ethnic groups, such as the Ashkenazi Jewish population; in this ethnicity there is a high population frequency (approximately 2%) of three founder mutations: BRCA1 185delAG, BRCA1 5382insC, and BRCA2 6174delT[31]. Kirchhoffet al[31] collected data from 586 Ashkenazi Jewish patients with pathologically confirmed diagnosis of CRC and from 5012 healthy subjects (controls), to assess whether having one of the three founder mutations could translate into an increased CRC risk. Mutations incidence in the two groups was matched and after adjusting for age at diagnosis and sex, it was observed that harbouring BRCA1/2 mutations was not associated with higher CRC risk[31].
Similarly, Nielet al[32], assessed whether positive breast cancer family history could represent a risk factor for developing other tumour types. The authors performed a case-control study comparing data from 1422 patients with pathologically confirmed CRC, coming from five major hospitals in northern Israel, with 1585 control subjects.They observed that neither BRCA1/2 founder mutations nor breast cancer family history were significantly correlated with increased CRC risk.
Another interesting topic to analyse is whether BRCA-related CRC could be characterised by peculiar clinico-pathological features. Xuet al[33] described CRC phenotype differences among suspected LS patients with MMR and BRCA/BRCA-like carrying variants. Among 22833 patients receiving radical surgery for cancer at Fudan University Shanghai Cancer Center, 202 underwent multigene testing covering 139 genes: 42 were carriers of a pathogenic MMR-gene variant, while 20 carried BRCA or a BRCA-like variant. The most relevant differences between the two groups of patients were that the mean CRC age at diagnosis was lower in the MMR group (44.95 yearsvs56.45 years); furthermore, BRCA/BRCA-like variants were associated with a significantly lower percentage of poorly differentiated CRC (5%vs33%) and lower metachronous CRCs. Moreover, tumour sidedness was different between the two groups, with more left sided colon cancer diagnoses in MMR-gene variants, whereas extra-CRC was significantly higher in BRCA/BRCA-like families. Finally, supposed LS patients carrying BRCA and BRCA-like variants had a longer progression free survival(PFS) compared with the others[33].
Based on histological and molecular features, CRC is actually a heterogeneous group of tumours, characterised by different prognosis and response to medical treatment. Mucinous carcinoma (MC) and adenocarcinoma (AC) are the two most common histological subtypes, accounting for 10%-15% and 85%-90% of cases respectively. In particular, MC is usually associated with worse prognosis: It is more frequently diagnosed in young patients and it is usually diagnosed later than AC, due to the late occurrence of symptoms; most of these tumours occur in the proximal colon.MC is also described as having quicker disease progression. Harpazet al[34] in their paper assessed the correlation between MC and BRCAm CRCs.
One thousand one hundred thirty-four mCRCs were analyzed: A statistically significant higher incidence of BRCA1/2 mutations in MC than in AC patients was observed (14.8%vs4.1%, respectively). Moreover, when considering the “mutation count”, defined as “somatic non-synonymous variants in encoding genes by exome sequencing” for each sample supplied by the database, they also observed that somatic BRCAm CRC has a higher mutation count than wild-type BRCA (BRCAwt); mucinous histology was also the strongest predictor of mutation count, independently of MSI as well as other variables too.
The authors conducted a prospective case-control study to confirm the results of their previous retrospective study: MC and AC CRC patients treated at Hadassah Medical Center in Jerusalem were prospectively enrolled. Thirty out of 53 MC cases were included, together with 40 controls: Authors observed a higher frequency of BRCAm patients in MC (40%vs27%, respectively,P= 0.2705 by chi-squared test). The difference was not statistically significant. Lack of concordance with their previous results might be due to the small cohort and to other limitations: In particular,mutation analysis revealed that variants frequently seen in MC histology such as KRAS, BRAF and PI3K mutations were not observed in this cohort of patients.However, analyzing the BRCA2m group, they noted a trend toward higher frequency of mucinous histology. Moreover, they confirmed the higher tumour mutational burden (TMB) in BRCAm compared to BRCAwt carriers.
While the effects of BRCAm are well known in females, studies focused on males are rarer; indeed, it has been hypothesised that male carriers of BRCA mutations are more likely to develop several types of malignancies, including melanomas, breast,prostatic and pancreatic cancer. However, clinicians used to test far fewer male than female patients for BRCAm, thus limiting our actual knowledge of clinic-pathological and prognostic implications of these kinds of tumours.
Sunet al[35] conducted a retrospective pan-tumour survey to identify tumour characteristics in BRCA1/2 male mutation carriers; clinical data of mutated male patients was compared with female patients carrying the same mutation, as well as BRCAwt male patients. Results of the analysis showed that CRC incidence was higher in BRCAm males than in BRCAm female patients and non-BRCAm males. In particular, they found that deleterious mutations in the BRCA2 gene were most frequent in CRC patients. Moreover, women carrying BRCA mutations have better overall survival (OS) and PFS than BRCAm males, principally due to the differences in tumour types and the therapeutic chances. Finally, males with deleterious BRCAm have increased OS compared to non-BRCAm.
Emerging therapeutic options in BRCA-mutated CRC
Five-years OS rate for patients with mCRC remains poor, around 14%. Standard chemotherapy for these patients includes 5-fluorouracil (5-FU) in association with irinotecan (FOLFIRI), oxaliplatin (FOLFOX) or both (FOLFOXIRI). These regimens are usually associated, particularly in first and second-line settings, with anti-EGFR or anti-VEGF targeted drugs, on the basis of tumour BRAF/RAS mutational status[36].
It has been previously described that BRCA1/2m cancers should have increased sensitivity to platinum compounds; on this basis, a few published case reports suggest that BRCA1/2 mutational testing, particularly in younger CRC patients might guide treatment selection in earlier settings of the disease. Soyanoet al[37] described the case of a young man with locally advanced rectal cancer [T3N1M0 according to tumournode-metastasis (TNM) staging]: He was tested for BRCA1/2 mutation and was found to be a BRCA1m carrier. After multidisciplinary evaluation, he started standard chemoradiotherapy with capecitabine as a radiosensitizer; however, when a BRCA1 mutation was found, oxaliplatin was added to standard chemoradiotherapy. It was also decided to opt for a total neoadjuvant strategy as in giving additional two cycles of chemotherapy (mFOLFOX6), after the end of chemoradiotherapy, just before surgery. The pathological report after surgery revealed complete pathological response.
Likewise, Linet al[38] described the case of a 25 years old man affected with locally advanced (cT3N2M0) rectal cancer who had an excellent response to oxaliplatin-based chemotherapy. The patient was due to receive neoadjuvant concurrent standard radiochemotherapy, but given the increased risk of radiation-induced infertility he refused any radiotherapy; 4 cycles of chemotherapy 5-FU and oxaliplatin-based (FOLFOX6)were then administered. After surgical primary tumour resection performed by laparoscopic total mesorectal excision (TME), the histological report showed complete response. Owing to this excellent but unexpected reaction to oxaliplatin-based treatment, the authors decided to perform next-generation sequencing in both DNA samples taken from the primary tumour sample and the peripheral blood sample;somatic BRCA2 variant was found, as well as high TMB.
This data suggest that the assessment of BRCA-status might be performed when a particularly deep response to platinum-based chemotherapy is reached.
It has already been explained how, in cells that harbour HR system mutations leading to defective DNA double-strand break repair, as in BRCAm patients, genome stability is maintained through the activity of BER and PARP enzymes. However,Pavioloet al[39] demonstrated that in PARPi-treated BRCA-defective CRC samples,even though DSBs are generated, they are not directly responsible for cell death.Indeed, more than DSBs, it was proven that rapid and anomalous repair of these defects, through means of DNA repair mechanisms different from HRD, leads to severe chromosomic aberrations which are the cause of cell death.
In clinical practice, several PARPi, such as Olaparib, Niraparib, Rucaparib, are widely used in treating ovarian, breast, pancreatic and prostate cancer harbouring BRCA mutations or HRD signature, and they are under investigation in many other neoplasms.
Veliparib (ABT-888) was the first PARPi to be studied in CRC treatment: Prior works demonstrated its efficacy in BRCA-deficient compared to proficient cells and proved that combining platinum-based compounds with PARPi provided increased response[40]. Harpazet al[34] described the use of Veliparib as maintenance therapy in a patient carrying a germline pathogenic BRCA1m, affected by metastatic KRAS and BRAF wild type rectal adenocarcinoma with pelvic and lung metastases. He underwent first-line chemotherapy with FOLFOX and Panitumumab, and obtained maximal response. Then he started maintenance veliparib, for as long as 2 years;treatment was then stopped after occurrence of new metastases.
Niraparib, a potent and selective PARP1/2i, has shownin vitroandin vivoefficacy in BRCA mutant cell lines; sensitivity to Niraparib monotherapy is 10-fold in BRCA mutant cell lines compared to BRCAwt cells[41].
A phase 2, multicentre, open-label study evaluating Rucaparib, an orally available PARP1i, in patients with unresectable, locally advanced or metastatic solid tumours with deleterious mutations in HR repair genes is currently still recruiting; the trial enrols patients with either somatic or germline variants and BRCA1 and BRCA2 mutations are allowed. (NCT04171700). Primary outcome is the best overall response rate[42].
In addition to that, a clinical trial evaluating the use of immune checkpoint inhibitors in solid tumours with BRCA mutation is still ongoing[43] (Table 1).
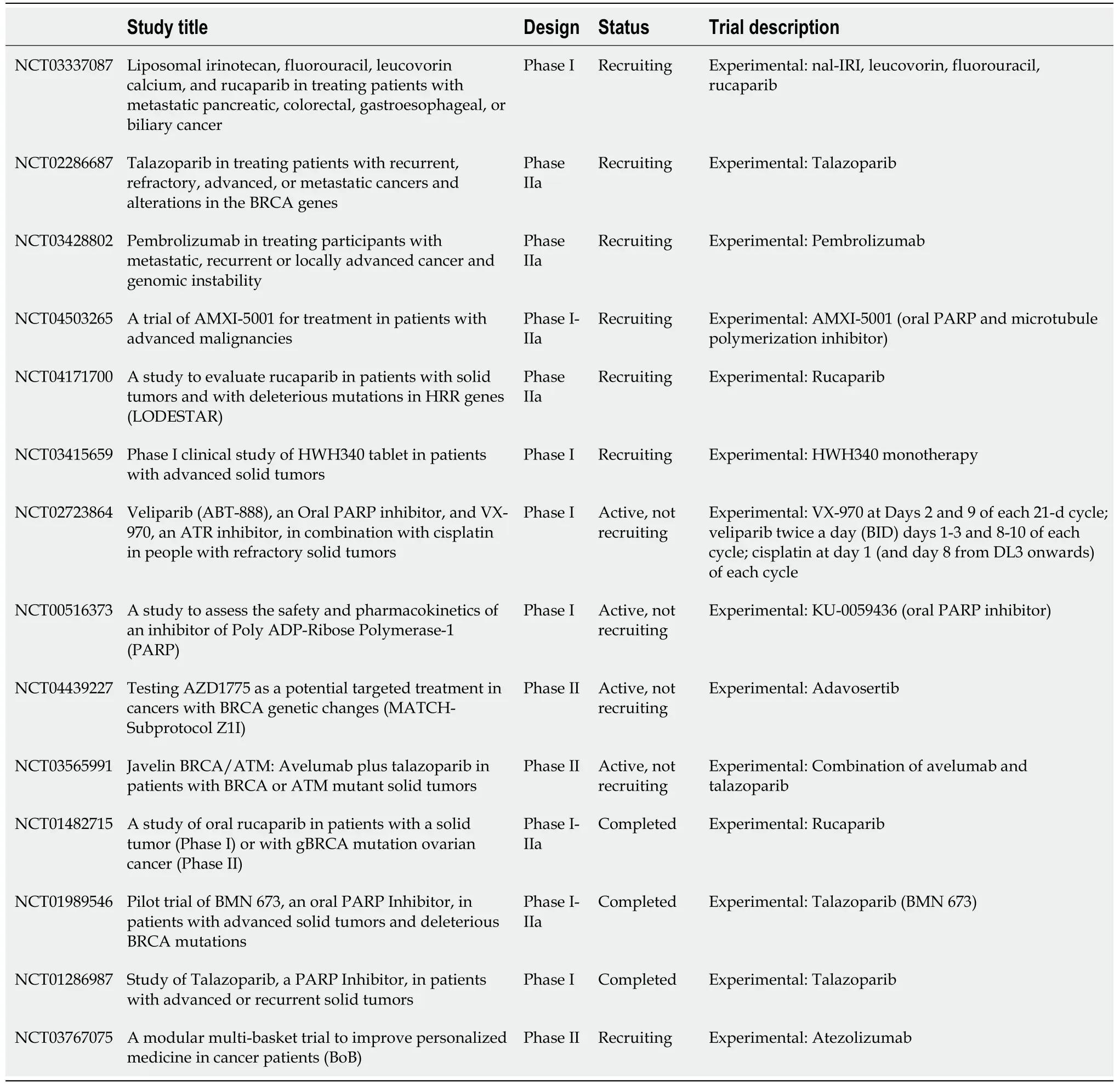
Table 1 Ongoing trials including patients carrying gastro-intestinal cancer with BRCA1m and/or BRCA2m and solid tumours with BRCA1m and/or BRCA2m
Despite these premises, at the moment there is still no evidence-based data that supports the use of these drugs in CRC patients; because of this it is suggested that PARPi-based clinical trials should be enforced in BRCAm carriers to further validate their use in this setting.
Furthermore, whole-genome sequencing in BRCA1/2m patients allowed identification of different signatures correlated with BRCA status; this BRCA-ness signature was present even in cells without BRCA1/2 mutations, suggesting that the signature might be more likely to predict HRD compared with simple assessment of BRCA mutational status[40]. Should the signature be more widely used, it can be expected that it will be tested to predict PARPi sensitivity.
GC
Worldwide, GC is one of the most common malignancies and the third leading cause of cancer-related mortality[44]. Approximately 8%-30% of patients have a positive family history[45], but only 1%-3% of GCs are truly hereditary[46]. GC predisposition has been linked to familial cancer syndromes, including hereditary diffuse GC[47], LS[48], Peutz-Jeghers syndrome[49], Li-Fraumeni syndrome[50], familial adenomatous polyposis syndrome[51], adenocarcinoma and proximal polyposis syndrome of the stomach[52].
The association between germline mutation of BRCA genes and risk of GC was investigated in several studies, although some aspects remain unclear.
Historically, carriers of germline BRCA1/2 mutations have shown a four to six-fold increased risk of developing GC compared to the general population[53,54] and the risk appears to be higher in patients with BRCA1 mutations than in patients with BRCA2 mutations[54,55].
Regarding BRCA2 carriers, the Polish study by Jakubowskaet al[56] assessed the importance of a family history of GC to predict the presence of BRCA2 mutations in ovarian cancer patients. In this study, BRCA2 mutation was found in 8 of 34 women with ovarian cancer and a family history of GCvs3 of 75 women with OR and a family history of ovarian cancer, but not of GC (OR = 7.4; 95%CI: 1.8-30;P= 0.004); this finding would confirm that GC is a BRCA2-related tumour.
Remarkably, a study conducted by the same group of authors, suggested that founder BRCA1 mutations reported in Polish breast/ovarian cancer patients do not contribute to increased GC risk[57].
Furthermore, Lorenzo Bermejoet al[58] demonstrated that GC before the age of 70 was twice as frequent in families with breast and ovarian cancers as in the general population; similarly, Schlebuschet al[59] found a high prevalence of GC in the BRCA2-positive families compared with the general population.
Nevertheless, in a study performed in the Netherlands, 139 BRCA2 families with 66 different pathogenic mutations were analysed and significantly increased risk of developing GC was not observed[16].
These conflicting data could be partially justified by the high prevalence of Ashkenazi Jews in the studies of Schlebuschet al[59] and Jakubowskaet al[56]; in fact,ovarian, pancreatic and gastric cancer as well as non-Hodgkin’s lymphoma have a higher incidence rate in the Ashkenazi population[60].
To further assess the impact of founder mutations in the Ashkenazi population,Figeret al[61] focused on a founder BRCA2 variant that is present in about 1.5% of the general Ashkenazi population and rarely in non-Ashkenazi Jews. In order to evaluate the contribution of this mutation to non-CRC GI cancer, they tested 70 consecutive unselected Ashkenazi Jews with GI malignancies that carried this specific mutation.They concluded that the rate of the Ashkenazi Jews with BRCA2 founder mutation in patients with GC was 5.7%, approximately five times higher than the general population, supporting the hypothesis of an increased risk of GC in BRCA2-carriers[46].
It has previously been reported that GC in BRCA2 carriers may be sex-related, as it seems to be more frequent in males[6,58]; in an interesting article, Cavanaghet al[62]argue that women carrying BRCA1/2 mutations usually develop early-onset breast and ovarian cancer and therefore may not survive long enough to develop GC at an older age; this would explain the high prevalence of GC (and other cancers) in male BRCA-carriers.
Regarding BRCA-associated tumours in males, Sunet al[35] recently conducted a retrospective pan-tumour survey on 346 cases of BRCA-associated tumours includingGC in males and a comparative analysis among male and female BRCA carriers (349 cases), as well as in male patients who were not BRCA carriers (4577 cases); similar incidences of BRCA mutations (6.0%vs6.6%) and age at diagnosis of BRCA-related tumour (median, 65vs60 years) were observed both in male and female patients.
Moreover, when evaluating both OS and PFS in patients who developed GC,interesting differences were observed: Compared with females, BRCA mutations in males were associated with decreased OS and PFS; however, subgroup analysis demonstrated that BRCA mutation was associated with increased OS in GC (HR for OS: 0.60P= 0.05). Considering the limited data about the outcome of gastric cancer in BRCA carriers, these results are of particular note. However, data regarding the prognostic impact of BRCA pathogenic variants in GC patients are still scarce.
Another study supporting these results was conducted by Halpernet al[63], where ten GC patients with BRCA mutations were assessed; 6/10 patients had metastatic disease. Median OS of all ten GC patients was 47.5 mo. Median OS for patients diagnosed with operable disease was 55.5 mo and was 32 mo for patients with metastatic disease. Particularly, patients with metastatic disease have a 1-, 2- and 3-year survival rate of 100%, 83.3% and 50%, respectively. Albeit the number of patients included in the analysis is rather limited, it is noteworthy, as it seems to support the idea that BRCA mutations might be associated with better survival: Nowadays median OS of patients with metastatic gastric cancer in western countries is usually around 10-12 mo.
In order to clarify the real incidence and outcome in BRCA patients with GC, a trial is currently active; in this prospective trial the investigators aim to evaluate the incidence of BRCA loss in patients with advanced GC and to observe the treatment outcome and the possibility of BRCA loss as a predictive and prognostic factor[64].
As previously discussed, existing clinical studies have suggested that some BRCAassociated tumours are sensitive to PARPi[18]. Therefore, a phase I/II study investigating side effects and best dose of lyposomal irinotecan and rucaparib when given together with fluorouracil and leucovorin calcium is currently available for BRCA patients with pancreatic, colorectal, gastroesophageal or respiratory cancer biliary.
Other basket trials available for patients with BRCA mutations are summarised in Table 1.
Finally, several cancers in BRCA carriers have been shown to be particularly sensitive to platinum-based chemotherapy[65,66], but studies evaluating the chemosensitivity of patients with BRCA germline mutations and GC are not available.
However, previous clinical and preclinical studies have reported an association between a low level of BRCA1 expression or a BRCA1 mutation and the chemosensitivity and prognosis of sporadic GC.
According to the American Joint Committee on Cancer, patients with a negative-BRCA1 sporadic GC are more likely to have a high grade of tumour, a high TNM stage and a poorly differentiated tumour[53,67]; at the same time, other studies have revealed that BRCAnegative sporadic GC are more sensitive to platinumbased adjuvant chemotherapy compared with BRCA1positive sporadic GC[68]. These findings indicate that patients with BRCA1negative GC have a longer OS time and improved prognosis, which suggests an important association between BRCA1 expression and platinumbased chemotherapy.
OEC
OeC is the seventh most common cancer and the sixth leading cause of cancer-related deaths worldwide[69]. The incidence rate of OeC varies considering the location;particularly high frequency is present in East Asia and Eastern/Southern Africa,where oesophageal squamous cell carcinoma (SCC) represents the main histology,while adenocarcinoma is more common in Western countries[70]. Most cases of OeC are sporadic and caused by somatic mutations[71] and oesophageal lesions are rarely described in hereditary CRC syndromes as LS or familial adenomatous polyposis(FAP). However, several studies reported the occurrence of OeC in patients with attenuated FAP or Gardner’s syndrome, which are both caused by mutations in theAPCgene[72]. The association between BRCA1/2 germline mutations and OeC has been explored in some studies.
Moranet al[73], in a family-based study, observed a relative risk increase of OeC(regardless of histology) (relative risk 2.9, 95%CI: 1.1-6) in families with BRCA1 mutations.
Conversely, BRCA2 carriers seem to have a higher risk of developing SCC than Adenocarcinoma.
In fact, contribution of BRCA2 mutations for the development of SCC has been reported in high- and low-risk Chinese populations, in the Turkmen population of Iran and in the very high risk region of northeast India. Specifically, Huet al[74] have reported five different BRCA2 mutations in 6 out of 44 (13%) patients with SCC in high-risk Chinese population, while Akbariet al[75] observed a nonsense BRCA2 pathogenic gene variant in eight SCC cases with a family history of OeC in the Turkmen population of Iran. Instead, Kaushalet al[76] have investigated 317 cases of SCC in a high-risk region of India and they conducted, performingBRCA2gene germline mutations, screening in 20 familial and 80 non-familial SCC patients: They found non-synonymous BRCA2 variants in 3 out of 20 patients with familial SCC,while no sequence alterations were found in 80 non-familial SCC cases. Moreover, a study to screen mutations ofBRCA2gene in 47 SCC patients from a low-risk Chinese population was conducted by Zhonget al[77]. They found 9 germline missense point mutations in apparently sporadic male patients, with a mutation frequency of 19%. In addition to that, when an additional cohort of 94 healthy controls underwent screening for the 9 mutations previously identified in SCC cases, only 2 positive individuals(mutation frequency 2%) were found.
Finally, a recent, large-scale study by Koet al[78] included 4517 individuals: 186 familial SCC patients from a high risk region of China, while the rest were healthy East Asian individuals (3289 Henan and 1228 moderate-risk Hong Kong Chinese). They identified BRCA2 Loss-of-function mutations in 3.23% (6/186) familial SCC patients compared to 0.21% in the East Asians (OR = 15.89,P= 2.48 × 10-10).
Regarding the therapeutic implications between BRCA carriers and OeC, few data are still available in the literature. However, encouraging preclinical studies would show that PARPi in combination with a DNA damaging agent might be beneficial in this setting[79]. Furthermore, the PARPi olaparib appears to sensitise OeC cells to fractionated proton irradiation[80].
DISCUSSION
Since the discovery in the mid-nineties of bothBRCA1/2genes and their impact on breast and ovarian cancer risk, both surveillance and risk-reducing surgical procedures have been developed as effective means to reduce the risk of developing both malignancies and to overall reduce the risk of death in carriers of these variants.However, recently, BRCA carriers have been recognised to be at risk of developing other kinds of cancer types, such as prostate, pancreatic cancer and melanoma[10-14].
GI cancers are a heterogenous group of malignancies that, taken together, represent the most common tumour type worldwide. Most tumours of this group are mainly due to environmental and lifestyle risk factors, while the weight of hereditary predisposition is rather low (usually around 5%-10% of all cancers). LS represents the most common form of hereditary predisposition to these kinds of tumours (mainly for CRC), while the role of BRCA1/2 mutations is less understood.
In our review we tried to summarize the current published evidence on the role of BRCA1/2 pathogenic variants in colorectal, gastric and OeC risk and prognosis.
As for CRC, there is no definitive consensus regarding increased risk of development of this malignancy: Some studies[17,29] seem to suggest that there might be an increased incidence of CRC in BRCA mutation carriers, particularly for BRCA1 carriers. In those studies where a positive correlation with increased risk of CRC development was proven, a trend towards younger age of onset was also suggested.On the other hand, other studies[30,31] have failed to confirm these findings, also in selected high-risk populations where the presence of BRCA variants is higher than in the general population (such as Ashkenazi Jews). Both these studies have some limitations that make direct comparison impossible: In the study of Phelanet al[17] the population consists entirely of female carriers, thus narrowing their findings to women. Conversely, the negative results of the studies of Kirchhoff and Bethany,conducted in the high-risk Ashkenazi Jewish population, might be influenced by the relatively higher risk of developing CRC in this ethnic group compared to people of other ethnicities[31,32,81].
While there is no consensus on the impact of germline BRCA1/2 mutations on CRC risk, their prognostic role is more clearly defined: A series of studies have shown how,in patients with CRC, BRCA mutations might increase the likelihood of response to chemotherapy. The evidence is, however, mainly derived from case reports or small retrospective case-control studies where patients are treated with oxaliplatin-based chemotherapy. All these studies suffer from selection biases, as only patients who had particularly significant responses to oxaliplatin-based treatment (and that should have more favourable prognosis), underwent genetic testing; there is a general lack of information concerning whether BRCA mutations may influence CRC prognosis regardless of treatment received. It is hoped that, in the near future, wide screening with multi-panel molecular testing in CRC patients will give us a clearer estimate of the real prevalence and prognosis of BRCA mutations; as multi-panel molecular testing will be used more frequently as a means to identify those patients who will be eligible to receive specific targeted drugs, and we will be able to identify more accurately people that might be carriers of germline BRCA mutations that would not be suspected on the basis of their family history.
The role of BRCA mutations in gastric cancer seems to be more clearly defined compared to CRC: A few studies have indicated an increased risk of gastric cancer in families with either BRCA1 or BRCA2 mutations. Even though few papers[54] suggest higher risk in BRCA1 carriers compared to BRCA2, most papers seem to agree that BRCA2 mutations are more clearly associated with an increased GC risk[56,58,59]. As well as in CRC, most studies that have shown a positive impact of BRCA mutations on GC risk have also shown an increased likelihood of early onset (< 70 years old) GC.
As stated before, all those studies that have enrolled specific ethnic groups that have a higher risk of harbouring BRCA mutations (such as Ashkenazi jews) have met with negative results; however, when the analysis was conducted by taking into account this factor, BRCA mutations maintained their causative role. Interestingly, most data that have focused on the prognosis of GC patients harbouring BRCA mutations agree on a positive prognostic role: This might partly be due to the fact that standard treatment of gastric cancer patients includes platinum compounds; it would be interesting to assess whether GC patients who harbour BRCA mutations and that do not receive any kind of platinum-based treatment (as those treated with fluoropyrimidines alone) retain the same positive prognosis.
Surprisingly, although OeC is usually considered as a cancer type where environmental and lifestyle factors predominantly influence the risk of the onset of disease, a lot of published evidence can be found concerning the role of BRCA mutations in this disease. Out of the two main histologies, SCC seems to be much more closely associated with BRCA mutations compared with adenocarcinoma. All papers seem to suggest that BRCA2 mutations are more frequently observed in this tumour type compared with BRCA1 mutations. Although studies that focused on the prognostic or predictive role of BRCA1/2 are lacking, it is expected that, based on the standard treatment options for this kind of disease that include platinum compounds and radiotherapy, more favourable outcome for patients who are carriers of these mutations can be expected.
The retrospective nature of these studies represents a major limitation; it is however interesting to notice that most of them indicate a positive correlation of germline BRCA1/2 mutations and an increased GI cancer risk. BRCA1/2 variant assessment will soon become one of the most promising fields of research in these kinds of diseases: Most of these tumours are treated with platinum compounds (either Oxaliplatin or Cisplatin) and it is expected that BRCA mutation carriers should have greater likelihood of response to treatment compared to wild type BRCA individuals.Furthermore, as in pancreatic cancer where PARPi have now become the standard care for patients with BRCA germline mutations[20], we can expect that the same might apply to other cancers arising from other areas of the GI tract: Indeed, PARPi are currently being evaluated in a wide number of clinical trials (Table 1).
In addition to that, it is actually recognised that, in CRC, GC and OeC, completely different clinical presentations are seen: While older patients have experienced in the last decade an improvement in survival (mainly due to introduction of screening procedures and advances in adjuvant therapy[36], younger patients are still those whose survival has not changed; most papers that have focused on BRCA1/2 mutations came to the conclusion that BRCA germline carriers are those who have the highest likelihood of early onset GI presentation. We believe that further research on early onset GI cancer might identify a greater number of patients where germline defects rather than environmental risk factors are the main causes of disease; this might also be associated with differences in treatment response. Moreover, it would advocate different surveillance procedures, as younger patients are not usually taken into account in most screening programmes.
CONCLUSION
In conclusion, we believe that this review has highlighted the importance of BRCA1/2 germline mutations as risk factors in GI malignancies and has drawn attention to future applications of this knowledge in clinical practice.
 World Journal of Clinical Oncology2021年7期
World Journal of Clinical Oncology2021年7期
- World Journal of Clinical Oncology的其它文章
- Esophagogastric junction adenocarcinoma: Preoperative chemoradiation or perioperative chemotherapy?
- Mechanisms of acquired resistance of BRCA1/2-driven tumors to platinum compounds and PARP inhibitors
- Therapeutic potential of thymoquinone in combination therapy against cancer and cancer stem cells
- Proteoglycans and their functions in esophageal squamous cell carcinoma