
纳米锗-锡/碳复合材料的合成与电化学性能研究
2021-07-31 06:25乔雪杨雪彪黄婷婷王新月贾子辰王红强
综合智慧能源 2021年7期
乔雪,杨雪彪,黄婷婷,王新月,贾子辰,王红强
(河北大学化学与环境科学学院,河北保定 071002)
0 引言
18世纪60年代以来,持续大量的化石燃料消耗造成了严重的环境污染,自然资源加速枯竭。为解决能源危机,人们研发了可高效转换和存储能量的电池技术。电池技术须具有低成本、可持续、安全性高和低毒性等优势[1-2]。经过20 多年的研发,锂离子电池成功实现商业化并且广泛应用于日常生活中[3-5]。目前商用锂电池主要采用插层型正极和石墨负极。正极材料包括LiFePO4(LFP)[6-7],钴和镍基氧化物(锂镍钴锰氧化物(NCM)),但其发展受限于较低的理论比容量(250(mA·h)/g),而石墨的理论比容量也仅为372(mA·h)/g[8],显著降低了锂离子电池体系的能量密度。为实现大规模应用,更小、更轻、更高的能量密度和可靠性是锂离子电池的发展趋势,而电极的开发和改性是研究的核心。
锗基负极材料因具有较高的理论比容量(1 600(mA·h)/g)而受到广泛关注,然而其作为锂离子电池负极应用却受到体积膨胀、导电性差等问题的限制。研究人员在合成纳米材料的基础上,通过结构调控和组分设计制备出具有优异电化学性能的负极材料。如Kwon 等[9]通过2 步法合成了双孔锗纳米结构,通过调整前驱体GeO2和SiO2的相对比例来形成不同种类的孔。这种双孔结构的锗负极在0.32 A/g 电流密度下充放电100 次后容量保持率为100.0%,在0.80 A/g 电流密度下循环300 次后容量保持率为94.4%。此外,多孔结构可缩短锂离子扩散路径,所以在8.00 A/g 大电流密度下电池比容量依然可以达到740(mA·h)/g,具有优异的倍率性能。Lin 等[10]通过熔融盐金属热反应法制备介孔空心锗微球,其比表面积为110 m2/g,平均孔径5.7 nm。该材料用作锂离子电池负极时,在0.32 A/g 电流密度下充放电150 次后仍具有1 291(mA·h)/g 的可逆容量,在1.28 A/g 电流密度下充放电循环400次后容量为1 217(mA·h)/g。说明介孔空心锗微球因其较小的颗粒尺寸及增大的比表面积,循环稳定性更好。Wang 等[11]通过模板法将锗量子点均匀嵌入三维有序多孔氮掺杂碳骨架中作为无黏结剂负极。在量子尺寸锗和多孔碳结构的共同作用下,锂离子和电子可被快速传输,并保持材料结构稳定以得到优异的循环性能和倍率性能。电池在5.00 A/g电流密度下充放电1 200 次后可逆容量超过1 000(mA·h)/g,每次循环容量衰减仅0.023%。
在合成纳米锗/碳材料中加入锡。锡的导电性远高于锗,可提高材料导电性,并与锂进行合金化反应。同时,锡嵌锂后膨胀率小于锗,可在一定程度上抑制体积膨胀。此外,锗与锡锂化反应的电位不同,锡锂化时锗可作为缓冲介质;锗-锡共锂化时,已经预锂化的锡可作为缓冲介质保护电极结构。这种逐步锂化过程可有效减轻电极在充放电时的体积应变,提高材料稳定性。
1 试验
1.1 材料制备
将200 mg 聚丙烯腈(PAN)加入10 mL 的N,N-二甲基甲酰胺(DMF)中,搅拌至PAN 完全溶解。加入293 μL 的GeCl4和15 mg 的SnCl2,继续搅拌至均匀后放入烘箱除去溶剂得到前驱物。将所得固体在氩气气氛下高温煅烧,先以2 ℃/min 的升温速率升温至250 ℃,保温4 h;再以5 ℃/min 的升温速率升温至700 ℃,保温2 h,得到C@GS3 负极材料。C@GS10制备方法与上述方法相同,GeCl4和SnCl2用量分别为260 μL 和47 mg。C@Ge 制备过程中GeCl4的用量为308 μL,不加入SnCl2。
1.2 材料表征
使用FEI公司的Nova Nano SEM450型扫描电子显微镜对材料进行形貌表征和能量分散光谱(EDS)元素分析。使用布鲁克公司D8 ADVANCE 型衍射仪对材料进行物相结构分析,射线源为Cu 靶(λ=1.540 56 Å),扫描范围为20°~80°。
1.3 材料电化学性能测试
将制得的材料与导电炭黑(Super P)及羧甲基纤维素钠(CMC)以8∶1∶1的质量比分散到蒸馏水中混成浆料,均匀涂覆在铜箔上,在120 ℃下真空干燥3 h。烘干后切成直径12 mm 的圆片称量备用。组装电池的全过程在充满氩气的手套箱里进行,电池型号为CR2032,所用电解液为1.0 mol/L 的LiPF6溶液,溶剂为含5.0%氟代碳酸乙烯酯(FEC)且体积比为1∶1∶1 的碳酸乙烯酯(EC)/碳酸二甲酯(DMC)/碳酸甲乙酯(EMC),隔膜为Celgard 2400聚丙烯膜。
用LAND CT2001A型电池测试系统对电池进行充放电测试,电压范围为0.01~1.50 V。循环伏安测试在Bio-Logic VSP 电化学工作站上完成,电压范围为0.01~1.50 V,扫描速率为0.1~1.0 mV/s。
2 试验结果与讨论
2.1 材料微观形貌表征
图1 为制备锗-锡/碳复合材料的流程。首先将PAN,GeCl4和SnCl2在DMF 中均匀混合,除去溶剂后将得到的前驱体进行高温煅烧,在此过程中PAN 在高温下发生碳化,氯化锗和氯化亚锡在高温和碳存在的条件下分别还原成锗和锡得到纳米锗-锡/碳复合材料。在锂化过程中,锡先与锂发生合金化反应,也就是预锂化,同时体积膨胀,未反应的锗充当缓冲剂并进行电子和离子传导。而后锡和锗共同锂化,此时LixSn充当缓冲剂缓解体积膨胀。整个过程中碳基质也起到了导电和适应体积变化的作用。
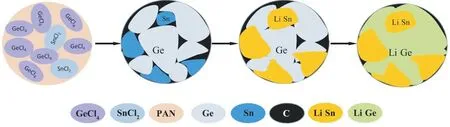
图1 锗-锡/碳复合材料合成示意Fig.1 Schematic illustration for the synthesis of Ge-Sn/C composite material
图2为C@GS3,C@GS10和C@Ge的扫描电子显微镜(SEM)成像图和EDS 分析结果。材料中存在C,Ge和Sn。没有Cl说明GeCl₄与SnCl2已完全反应。表1 为C@Ge,C@GS3 和C@GS10 中C,Ge 和Sn 的原子 百 分 比。计 算 得 出C@GS3 和C@GS10 中,Sn 占Ge和Sn总量的摩尔分数分别为1.5%,5.3%。
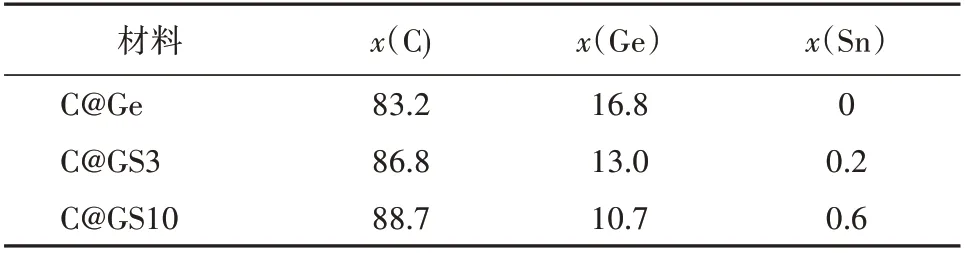
表1 C@GS3,C@GS10,C@Ge中C,Ge,Sn原子百分比Tab.1 Atomic percentage of C,Ge and Sn in C@GS3,C@GS10 and C@Ge %

图2 C@Ge,C@GS3和C@GS10的SEM结果和对应的EDS结果Fig.2 SEM images and corresponding EDS images of C@Ge,C@GS3 and C@GS10
2.2 材料物相结构表征
图3 为C@GS3,C@GS10 和C@Ge 复合材料的X射线衍射(XRD)图。由图3可见,3种材料都具有锗衍射峰,且与立方锗(JCPDS No.04-0545)的特征峰相对应[12-14]。
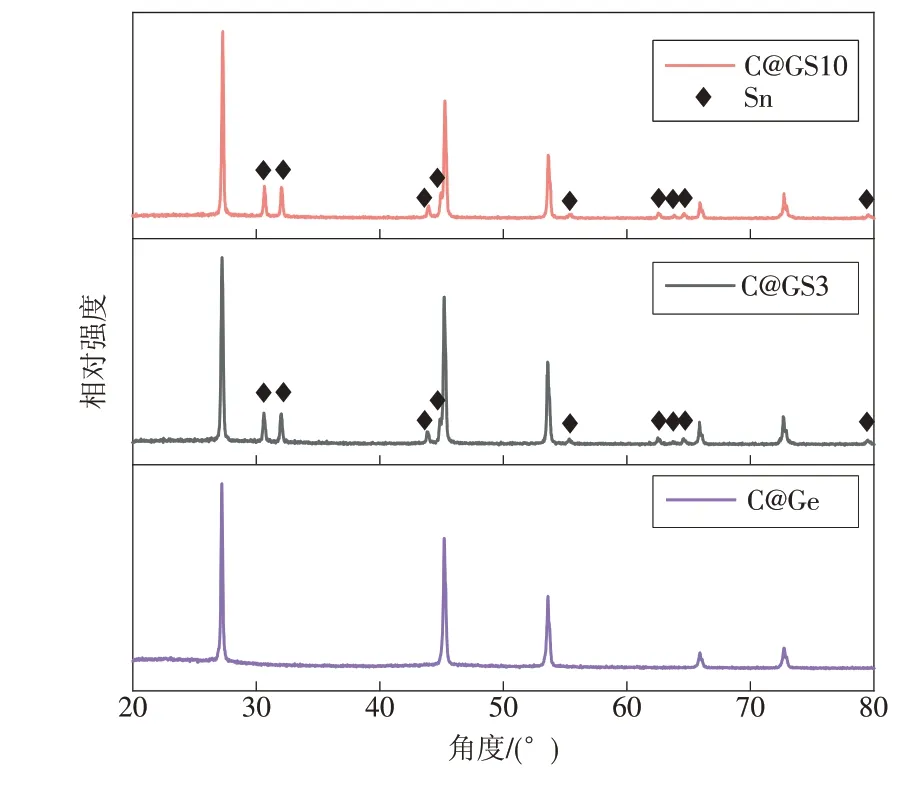
图3 C@GS3,C@GS10和C@Ge的XRD结果Fig.3 XRD patterns of C@GS3,C@GS10 and C@Ge
C@GS3 和C@GS10 中存在相对较弱的锡特征峰,这与元素分析图中结果一致。材料中均不存在反应物氯化锗和氯化亚锡,说明已经反应完全。而图中没有与碳相关的衍射峰,是因为碳的特征峰与强度较高的锗(111)晶面衍射峰位置接近,2 种衍射峰发生重合,导致碳的衍射峰不容易被观察到。
2.3 材料电化学性能分析
图4a 为C@GS3,C@GS10 和C@Ge 在0.50 A/g电流密度下的循环性能。由图4a 可见C@GS3 的初始放电容量最高,为1 292(mA·h)/g,而C@GS10,C@Ge 的容量分别为1 256,1 053(mA·h)/g,这是因为加入少量的锡后提高了材料的导电性,被碳包裹的锗反应更加完全,而C@GS10 的容量较低是因为锡的理论容量低于锗,含量增加后会降低材料整体的容量。3 个电极的首次循环库伦效率都在65.0%以下,这种不可逆的容量损失是因为电极材料与电解液在固液相界面上发生反应,形成了电子绝缘的固体电解液相界面膜(SEI)[15-17]。持续充放电100次之后,C@GS3 还具有510(mA·h)/g 的容量,相当于第2 次循环容量的65.0%,而C@GS10 和C@Ge 相对于第2 次循环的容量保持率分别为64.0%和36.0%,说明加入锡之后材料的循环稳定性有明显的提升。但是随着锡含量的增多,C@GS10 的比容量整体偏低。
图4b 为C@GS3,C@GS10 和C@Ge 在0.20,0.50,1.00,2.00,5.00,8.00 和10.00 A/g 电流密度下的倍率性能。由图4b 可见C@GS3 在不同电流密度下都具有最高的容量,在10.00 A/g的大电流密度下仍然具有460(mA·h)/g 的容量。而C@GS10 因为锡含量增多而导致容量较低,C@Ge 虽然在首次循环时具有较高容量,但是随着循环次数增多衰减严重,在10.00 A/g 电流密度下只有210(mA·h)/g的容量。当电流密度恢复至0.20 A/g 时,C@GS3 的容量为670(mA·h)/g,C@GS10,C@Ge 的容量分别为580,520(mA·h)/g,而且由图4a 及图4b 可见,C@GS3 和C@GS10 在经过70 次充放电后稳定性要高于C@Ge。结果表明,锡的加入不仅提高了材料的结构稳定性,而且可以改善其倍率性能。
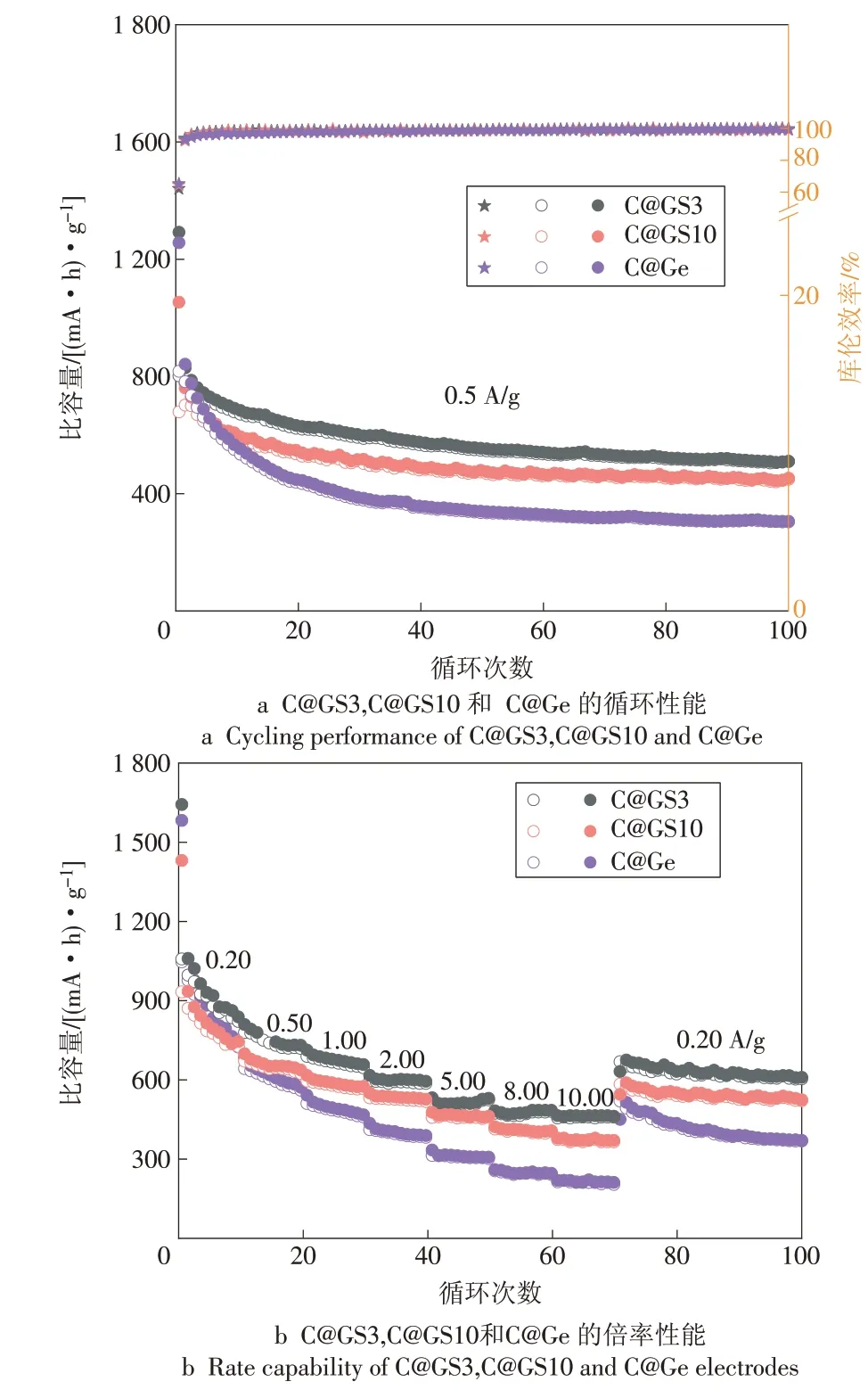
图4 C@GS3,C@GS10和C@Ge的循环性能和倍率性能Fig.4 Cyclical and rate performances of C@GS3,C@GS10 and C@Ge
图5为C@GS3,C@GS10和C@Ge在不同电流密度下的充放电曲线,图中标注的是在10.00 A/g电流密度下充电和放电的中值电压差-极化电压,分别为0.40 V,0.44 V和0.52 V。
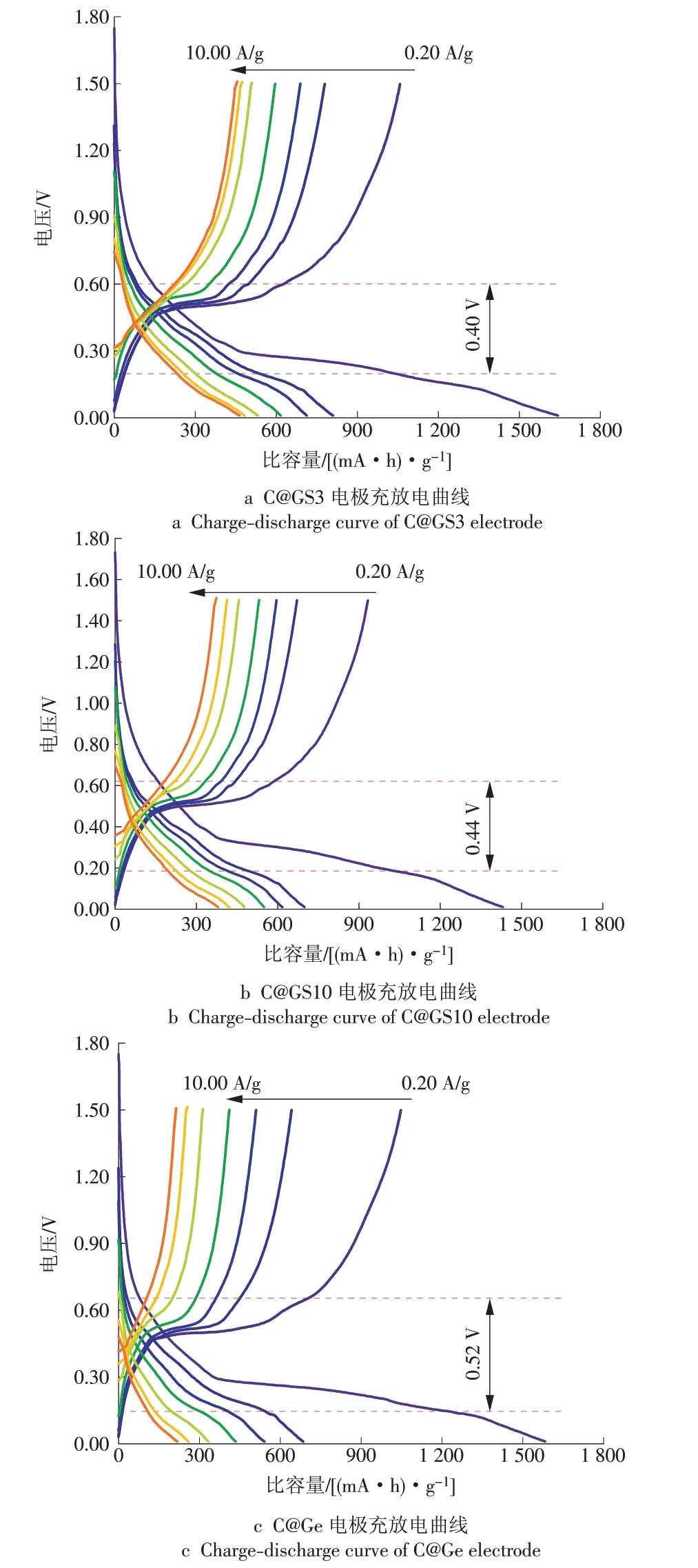
图5 C@GS3,C@GS10和C@Ge电极的充放电曲线Fig.5 Discharge/charge curves of C@GS3,C@GS10 and C@Ge electrodes
表2 为3 种材料在其他电流密度下的极化电压。由表2 可见,C@GS3 和C@GS10 的极化电压小于C@Ge,说明加入锡可有效缓解电极的极化现象,增强电子传导和离子扩散,提高电极反应动力学性能[18-20],这也是C@GS3具有优异倍率性能的原因。

表2 不同电流密度下C@GS3,C@GS10和C@Ge的极化电压Tab.2 Polarized voltages of C@GS3,C@GS10 and C@Ge electrodes under different currency V
图6为C@GS3,C@GS10和C@Ge在较大电流密度2.00 A/g 下的长循环性能。在前2 次0.50 A/g 的小电流活化之后,C@GS3 和C@GS10 表现出优异的循环性能。循环400 次后,C@GS3 和C@GS10 的容量分别保持在386,310(mA·h)/g,而C@Ge 的容量只有205(mA·h)/g。C@GS3 和C@GS10 在大电流密度下依然具有优异的循环稳定性,得益于锡组分的加入,不仅增强了锗的导电性,而且有效抑制了锗的体积膨胀,保证了电极结构的稳定。
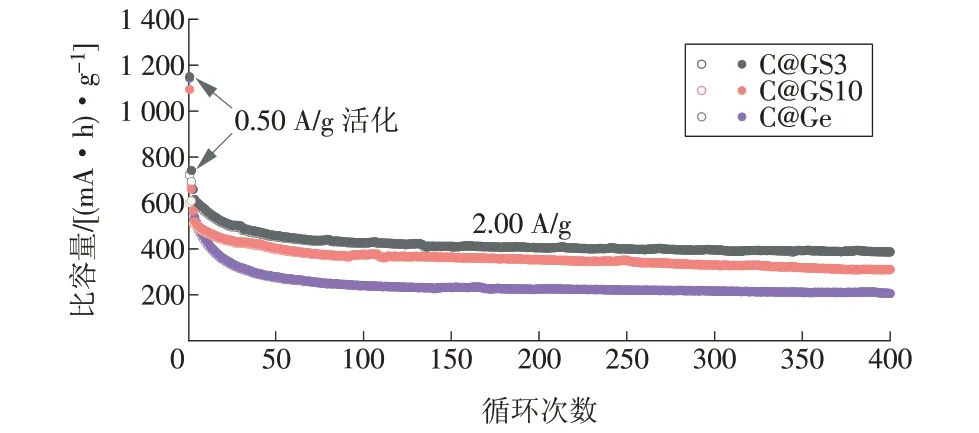
图6 C@GS3,C@GS10和C@Ge在2.00 A/g电流密度下的长循环性能Fig.6 Long cyclical performances of C@GS3,C@GS10 and C@Ge electrodes at a current density of 2.00 A/g
图7 为C@GS3 和C@Ge 在0.2,0.4,0.6,0.8,1.0 mV/s扫描速率下的循环伏安曲线。
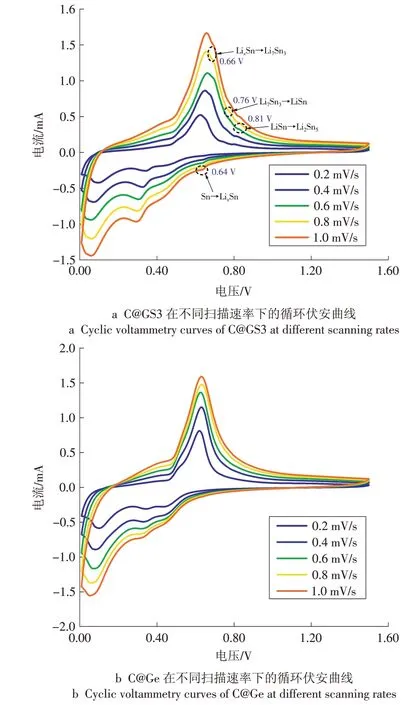
图7 C@GS3和C@Ge在不同扫描速率下的循环伏安曲线Fig.7 Cyclic Voltammetry measurements of C@GS3 and C@Ge at different scanning rate
由图7 可见,与C@Ge 相比,C@GS3 不仅有锗的氧化还原峰[21-22],而且有电流强度较弱的锡的氧化还原峰。放电过程中在0.64 V锡预锂化成LixSn,而后是锗、锡的共锂化;在充电过程中,先是锂化的锗脱锂,然后锂化锡逐步脱锂。循环伏安曲线表明,C@GS3 材料是经过逐步锂化反应的。在锡嵌锂时,未反应的锗作为缓冲介质,保护整个电极结构不被破坏。在接下来的共锂化过程中,预锂化的锡可以缓解体积膨胀,从而保证了电极材料的完整性[23-24]。
图8为C@GS3,C@GS10和C@Ge的电化学交流(EIS)阻抗图谱,图中中频区域对应电荷在界面的传递电阻。由图8可见,C@GS3的电荷转移电阻最小,表明加入锡可促进电荷传导,提高材料导电性。

图8 C@GS3,C@GS10和C@Ge的EIS阻抗图谱Fig.8 Electrochemical Impedance Spectroscopy of C@GS3,C@GS10 and C@Ge
3 结论
为解决锗负极电导率低、循环稳定差等问题,在锗/碳材料中加入锡,明显改善其电化学性能。先将锗与无定形碳进行复合,提高锗导电性并可适应锗体积变化。锡的导电性优于锗,加入锡可进一步提升材料导电性。而且锡可与锂发生反应,又与锗锂化电位不同,放电过程中预锂化的锡可在锗-锡共锂化时充当缓冲介质,保护电极结构。C@GS3 和C@GS10 具有良好的循环稳定性。在0.50 A/g 电流密度下循环100 次后,C@GS3 仍具有510(mA·h)/g的容量,相当于第2次循环时容量的65.0%。
猜你喜欢
纺织科学研究(2021年7期)2021-08-14
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
中国有色金属学报(2018年2期)2018-03-26
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
中国塑料(2016年3期)2016-06-15
浙江大学学报(工学版)(2016年9期)2016-06-05
电源技术(2015年5期)2015-08-22
电源技术(2015年11期)2015-08-22
