La掺杂CaMnO3的合成及其对木质素热解的催化性能*
2021-07-28 10:06陈彦广周春宇韩洪晶王海英王程昊王怡真关金双
化工科技 2021年3期
陈彦广,周春宇,韩洪晶,王海英,王程昊,王怡真,关金双,宋 华
(1.东北石油大学 化学化工学院,黑龙江 大庆 163318;2.黑龙江省石油与天然气化工省重点实验室,黑龙江 大庆 163318)
木质素是地球上含量最丰富的芳香族天然高分子聚合物,是一种来源广泛、成本低廉的可再生资源[1]。在中国,每年制浆造纸和制糖工业会产生大量的木质素,其中大部分用于直接燃烧获取热量,不但浪费资源还会引起环境污染。由于木质素结构复杂,物理性质稳定,直接转化利用比较困难,只有在高温加热或者催化作用下(如热解、催化水解、催化加氢、催化氧化和酶催化转化)才能将木质素解聚生成燃料油或化学品等,以实现木质素的资源化和高值化利用,进而替代部分化石燃料,对经济的可持续发展和环境保护具有重要意义[2-4]。
酶催化转化条件比较苛刻,因此目前木质素转化主要采取催化水解、催化加氢、催化氧化等方法。韩洪晶等[5]以La掺杂镁铝水滑石(LaMgAl-LDHs)为前驱体,通过焙烧过程调控制备纳微尺度LaMgAlOx固体碱氧化物,在乙醇-水体系中对木质素磺酸钙进行催化解聚,液相收率达46.26%,产物主要包括苯酚类和愈创木酚类,但其分离工序较长。杨茂林[6]采用浸渍法制备不同钌负载量的Ru/γ-Al2O3催化剂,并在氢气初始压力为3 MPa,t=250 ℃对木质素进行催化加氢实验,t=8 h木质素的转化率达到76.2%,单酚类收率达到4.63%。欧阳新平等[7]采用H2O2和CuO/FeSO4复合氧化体系,在180 ℃微波辅助条件下对木质素进行了氧化降解实验,木质素转化率高达90.88%,其中紫丁香酚类化合物的选择性可达11.86%,但整个工艺过程复杂,成本较高。陈彦广等[8]采用固相法制备了CaZr0.2Fe0.8O3催化剂并利用其对甘蔗渣木质素(BL)进行了催化热解实验,液相产物收率为23.71%,主要为苯酚类、紫丁香酚类、邻苯二酚类和愈创木酚类,其余为苯类和醚类化合物,但产物中各酚类化合物选择性较难调控。
钙钛矿氧化物具有良好的氧离子和电子传导性能,其结构为ABO3,其中A通常为碱土金属元素或稀土金属元素,B为半径较小的过渡金属元素[9]。钙钛矿氧化物由于具有氧离子和电子传递性好、化学和热稳定性高等特点,可用于木质素的催化热解[10]。作者利用溶胶-凝胶方法合成了CaMnO3,并通过在A位掺杂稀土金属元素La,合成了一系列La掺杂的CaMnO3,并对其催化热解木质素性能进行了评价。
1 实验部分
1.1 试剂与仪器
BL:化学纯,中国科学院广西植物研究所;柠檬酸(CA)、乙二胺四乙酸(EDTA)、四水硝酸钙、硝酸锰、六水硝酸镧、浓氨水:分析纯,天津市大茂化学试剂厂;高纯氮气:纯度大于99.999%,大庆市雪龙石化技术开发有限公司。
马弗炉:ZY-MB,洛阳高新开发区中苑实验电炉厂;粉末压片机:769YP-15A,天津科器高新技术公司;电子分析天平:FA124,上海海康电子仪器厂;恒温油浴锅:ZNCL-GS240×150,巩义市予华仪器有限责任公司;超声波清洗器:JP-040ST,上海易净超声波仪器有限公司;远红外线快速恒温干燥箱:HY,上海迈捷实验设备有限公司;高压固定床微反应装置:JQ-16,海安石油科研仪器有限公司; X射线衍射仪:D8 VENTURE,德国Bruker/AXS公司;电子显微镜:JSM-6360LA,日本电子株式会社(JEOL);比表面积分析仪:NOVA 2000e,美国Quantachrome Instruments公司;热重分析仪:HTG-1,北京恒久实验设备有限公司;气相色谱-三重四级杆质谱联用仪:Aglient 7890-7000,美国Agilent公司。
1.2 表征手段
利用X射线衍射仪(XRD)对钙钛矿产物晶相进行表征分析,以Cu靶Kα为辐射源,波长λ=0.154 06 nm,管电压为45 kV,电流为30 mA,扫面范围2θ=10°~80°,扫描速率为5°/min;利用电子显微镜(SEM)对固体样品的形貌进行表征分析,加速电压为5.0 kV,放大倍数为1×104~1×105倍;利用比表面积分析仪对钙钛矿产物的比表面积进行表征分析;利用热重分析仪对干凝胶进行热重分析(TG)和微分热重分析(DTG),对钙钛矿干凝胶热力学性能评价;利用气相色谱-三重四级杆质谱联用仪(GC-MS)对木质素热解液体产物进行分析,色谱柱型号为DB-5 ms UI 30 m×0.25 mm×0.25 μm,进样量为0.5 μL;进样口温度为280 ℃;进样口模式:不分流;色谱柱流量模式:恒流,流速为1.0 mL/min;柱箱升温程序为40 ℃(保持3 min),以5 ℃/min升温至300 ℃;传输线温度为300 ℃;四极杆温度为150 ℃;采集模式:全扫描m/z=40~550;溶剂延迟4.5 min。
1.3 Ca1-xLaxMnO3(CLM-x)的制备
将n(CA)∶n(EDTA)=1.6∶1溶液加入到120 mL去离子水中搅拌均匀,搅拌过程中逐滴加入浓氨水至溶液透明澄清;按照n(CA)∶n(EDTA)∶n(Ca2++Mn2++La3+)=1.6∶1∶1依次加入四水硝酸钙、硝酸锰溶液和六水硝酸镧超声分散30 min至完全溶解,加入氨水调节体系pH值;将上述溶液置于80 ℃油浴锅中恒温5 h至凝胶状态,将凝胶置于120 ℃干燥箱中干燥12 h;最后在800 ℃马弗炉中焙烧6 h,自然冷却至室温得到CLM-x。
1.4 催化性能及稳定性的评价
称取m(CLM-x)∶m(BL)=1∶3粉末放入研钵中研磨使其混合均匀,p=20 MPa用压片机压10 min得到CLM-x与BL的混合压片,然后将其破碎成约1.7 mm颗粒,加入到高压固定床微反应装置中,以50 mL/min的氮气为载气,以20 ℃/min升温至650 ℃,恒温反应2 h,液体产物经冷凝收集进行GC-MS分析。反应后的CLM-x因结焦失活,在马弗炉中800 ℃通入空气焙烧6 h实现再生。
2 结果与讨论
2.1 La掺杂量对CLM-x晶相和形貌的影响
不同La掺杂量下锰酸钙的XRD谱图见图1。
由图1可知,5种产物在2θ=22.73°、32.77°、40.50°、47.06°、58.52°、68.83°和78.23°出现CLM-x钙钛矿的衍射特征峰,说明Ca、Mn和La完全进入钙钛矿骨架中,此时钙钛矿呈立方晶相[11]。随着La掺杂量的增加,钙钛矿的特征峰向左偏移,2θ值减小,这是因为通过在A位掺杂原子半径较大的La会使晶胞参数变大,晶面间距变大进而使衍射特征峰向低角度位移[12];掺杂量x=0.5,特征峰的强度最大,说明此时钙钛矿的结晶度最高,因此选取x=0.5为最佳掺杂量。
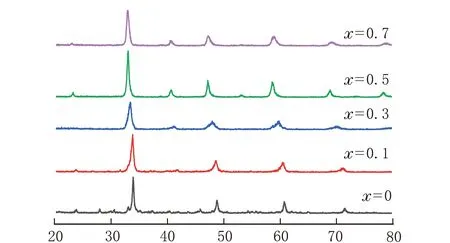
2θ/(°)图1 不同La掺杂量CLM-x的XRD谱图
不同La掺杂量锰酸钙的SEM照片见图2。

a x=0.1

b x=0.5

c x=0.7图2 不同La掺杂量CLM-x的SEM照片
由图2可知,x=0.1,钙钛矿晶粒边界模糊发生黏连和团聚现象;x=0.7,合成的钙钛矿发生轻微的黏连,颗粒大小不一且粒径尺寸较大;x=0.5,钙钛矿呈现疏松的块状结构,此时钙钛矿粒径较小且孔结构丰富。
2.2 前驱体pH值对CLM-0.5的影响
不同前驱体pH值下所制备的CLM-0.5的XRD谱图见图3。
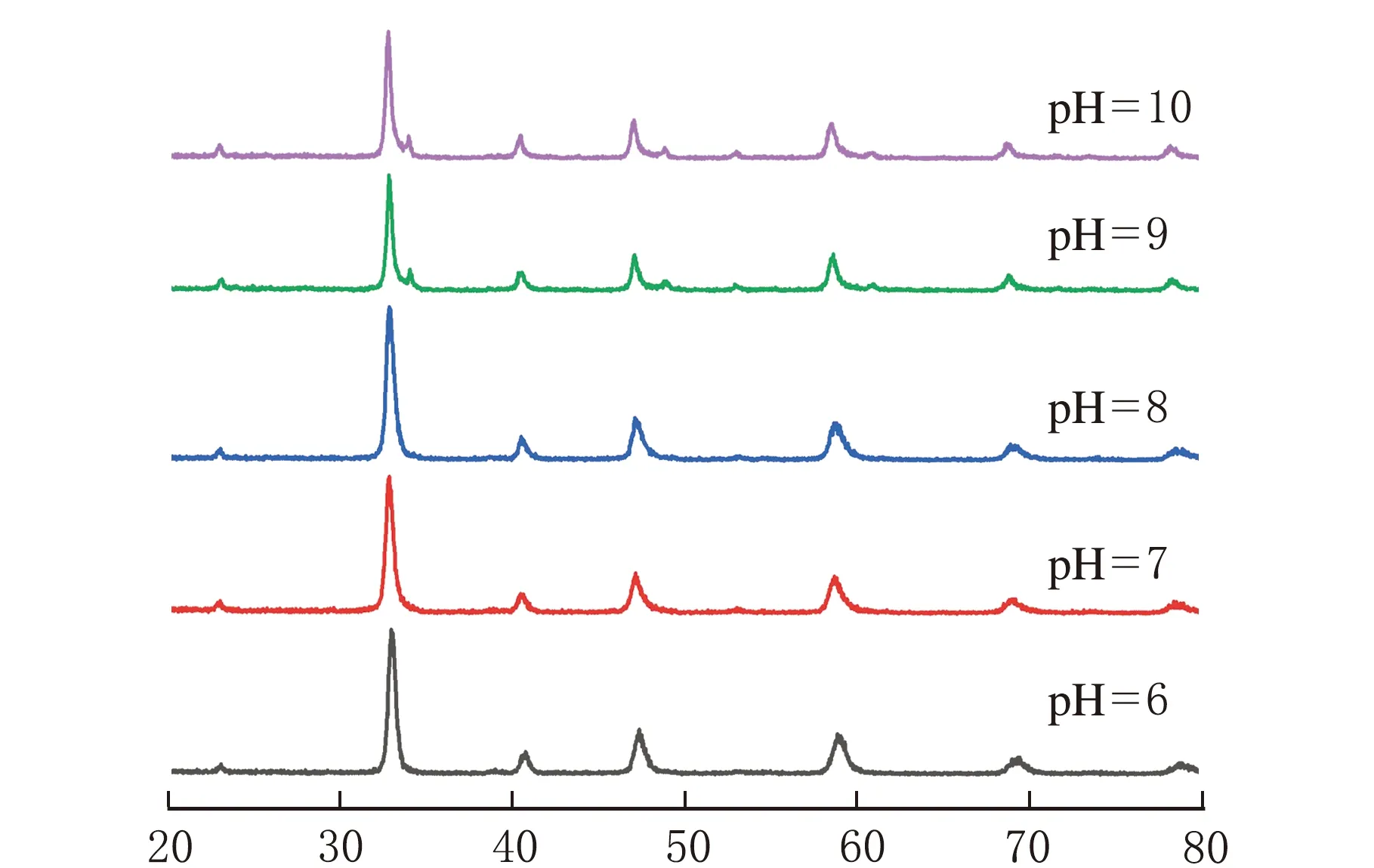
2θ/(°)图3 不同前驱体pH值合成的CLM-0.5的XRD谱图
由图3可知,所合成5个样品均存在明显的钙钛矿衍射特征峰,随着pH值的增大,钙钛矿特征峰的强度先增大再减小,并伴随杂峰的出现,前驱体pH=8,特征峰强度达到最大,此时结晶度最高。这是由于柠檬酸在不同pH值下的水解程度不同,pH=6,柠檬酸水解不完全,形成的络合物不稳定,故焙烧后钙钛矿的结晶度较低;pH=8,柠檬酸解离程度最大,形成的金属络合物较稳定,有利于钙钛矿的形成;随着pH值继续增加造成体系中的OH-含量增加,此时金属离子和OH-进行反应生成氢氧化物沉淀,造成柠檬酸络合物减少,不利于钙钛矿的形成,并伴随杂质的生成[13]。
前驱体pH=6、8、10合成CLM-0.5的SEM照片见图4。
由图4可知,前驱体pH=6,合成的钙钛矿颗粒堆积比较密集;pH=10,合成的钙钛矿样品平均晶粒尺寸增加、呈片状结构,大小不均一;pH=8,合成的钙钛矿颗粒呈规则的粒状结构,并且颗粒尺寸较小分布十分均匀,锰酸钙镧晶相和形貌相对较好。综上所述,选择前驱体pH=8。

a pH=6

b pH=8

c pH=10图4 不同前驱体pH值合成的CLM-0.5的SEM照片
2.3 焙烧气氛对CLM-0.5晶相的影响
不同焙烧气氛下得到CLM-0.5的XRD谱图见图5。
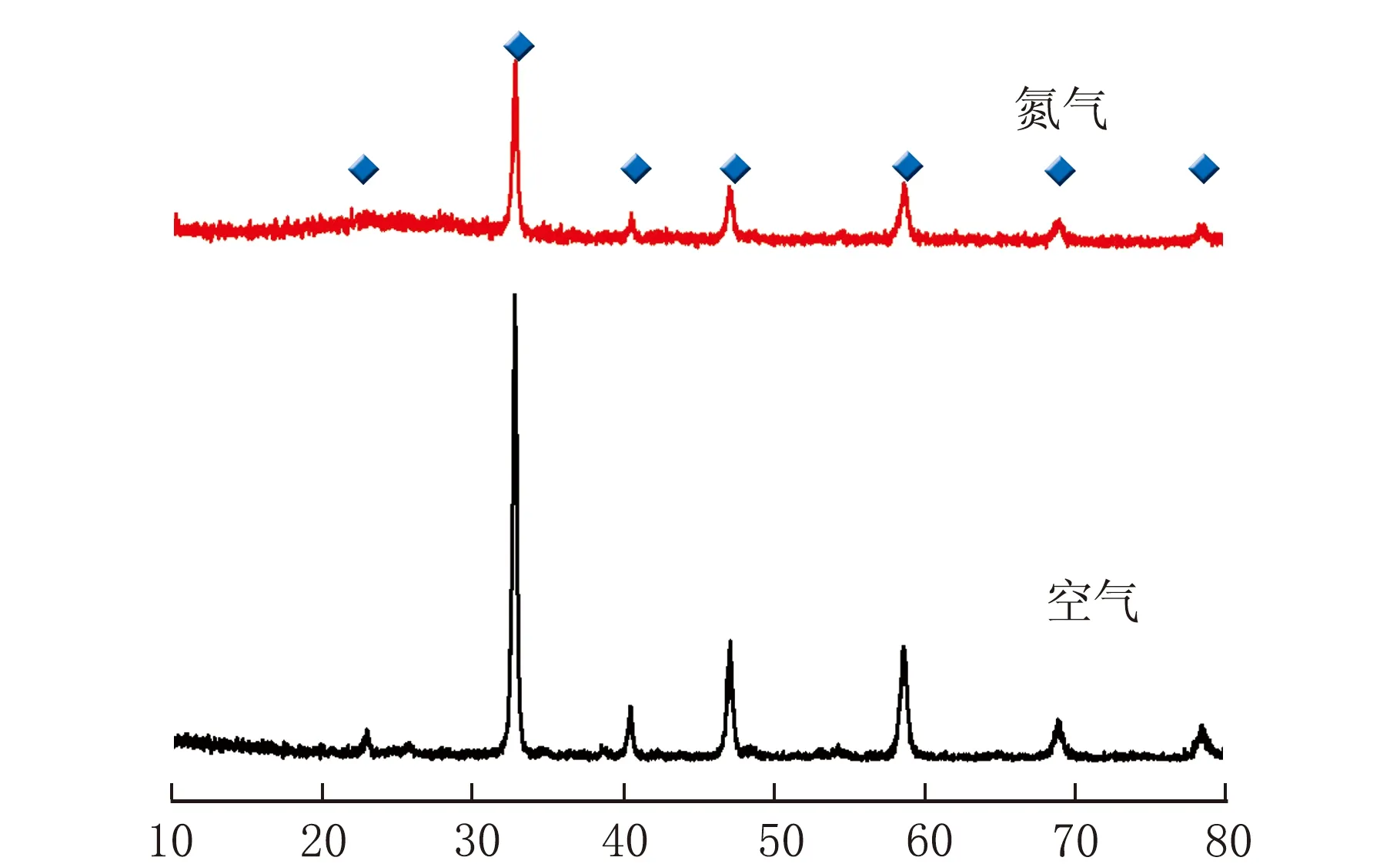
2θ/(°)图5 不同焙烧气氛合成的CLM-0.5的XRD谱图
由图5可知,在焙烧过程中,通入空气合成的CLM-0.5与通入氮气所合成的CLM-0.5相比,前者的衍射特征峰更明显,峰型尖锐,说明合成的产物结晶度更高。通入氮气合成的CLM-0.5存在金属氧化物杂峰,可能是由于Ca(NO3)2·4H2O、Mn(NO3)2和La(NO3)3·6H2O分解为氧化物后未能完全发生反应生成钙钛矿结构[14]。在通入空气焙烧时,作为络合剂的柠檬酸和乙二胺四乙酸发生氧化反应,放出大量热,并产生二氧化碳,从而使样品自身发生破碎,增大比表面积,使得样品的晶型更加完整。而通入氮气时,柠檬酸和乙二胺四乙酸只能在设定温度下自行分解,对体系形成的促进作用远低于通入空气时的情况,甚至可能产生杂质,影响产物纯度。因此,选取空气气氛为宜,在该气氛下制备的CLM-0.5晶相较好。
2.4 焙烧温度对CLM-0.5晶相的影响
CLM-0.5干凝胶的热稳定性能分析TG-DTG曲线见图6。
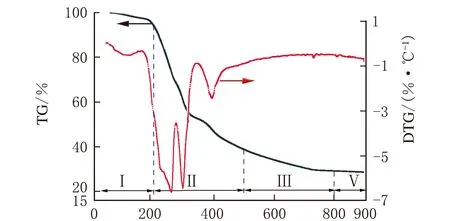
t/℃图6 CLM-0.5干凝胶的TG-DTG曲线
由图6可知,失重主要分为4个阶段。第一阶段由室温至200 ℃,此阶段失重率约为5%,主要是干凝胶吸附的氨气和水的脱除;第二阶段为200~500 ℃,该阶段失重率约为55%,在250 ℃和300 ℃有2个较大的失重峰,其原因是干凝胶中的络合剂与金属硝酸盐发生了分解;第三阶段为500~800 ℃,该阶段失重较缓慢,是中间产物逐渐发生分解,钙钛矿晶核逐渐生成的过程;第四阶段发生在800~900 ℃,在这一阶段基本不发生失重变化,这是因为CLM-0.5晶核已经形成,因此钙钛矿适宜焙烧条件为800 ℃。
不同焙烧温度合成CLM-0.5的XRD谱图见图7。
由图7可知,焙烧温度为600 ℃,未出现钙钛矿的衍射特征峰,随着焙烧温度升高出现钙钛矿的特征峰并且强度逐渐增强,焙烧温度达到800 ℃,钙钛矿的特征峰强度达到最大,说明此时结晶度最高,随着温度继续升高,特征峰呈下降趋势。这是由于高温焙烧过程中,温度升高有利于钙钛矿晶粒连续结晶,但是温度过高容易导致钙钛矿颗粒烧结。
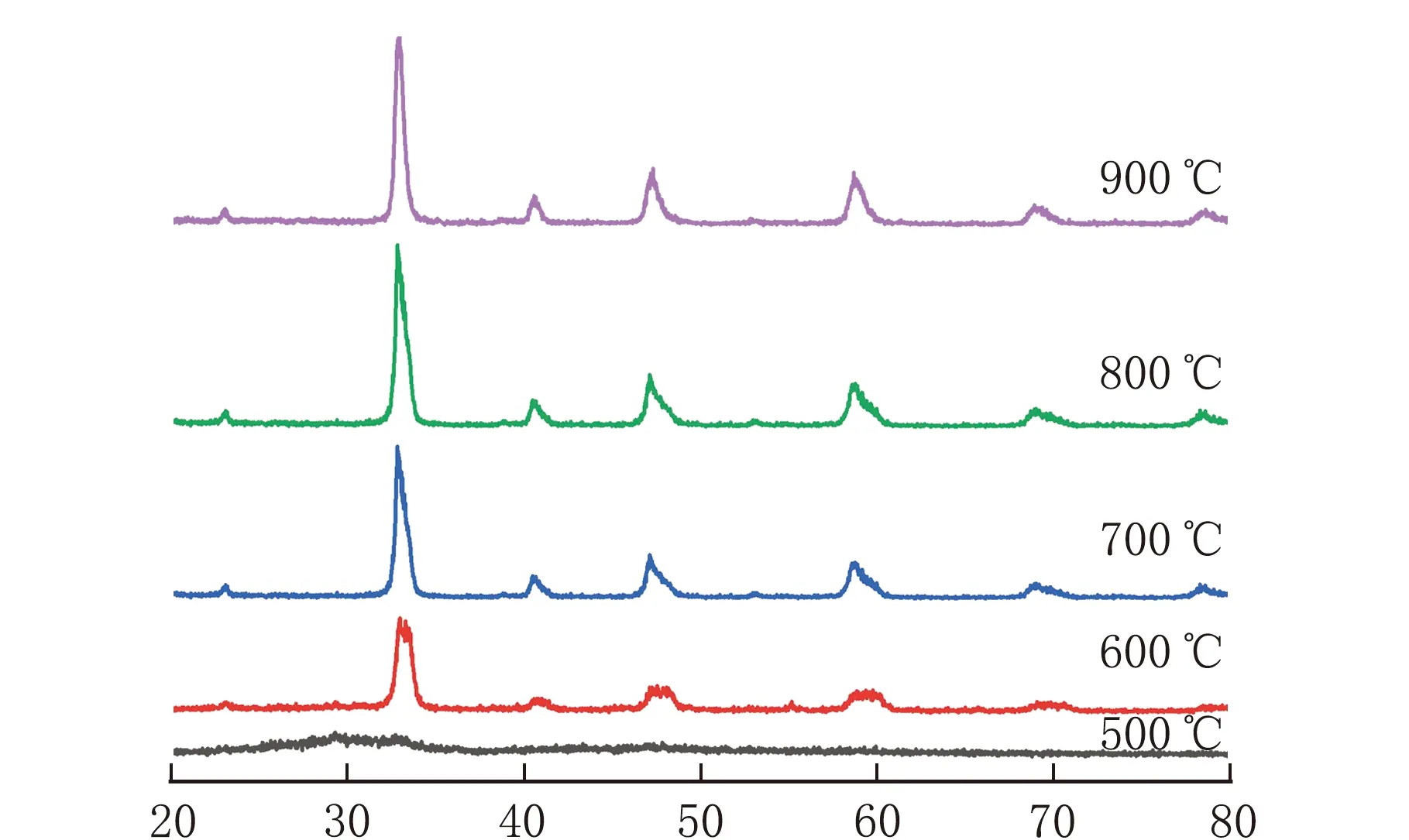
2θ/(°)图7 不同焙烧温度合成CLM-0.5的XRD谱图
不同焙烧温度合成的CLM-0.5SEM照片见图8。
由图8可知,700 ℃合成的钙钛矿颗粒大小不均一,800 ℃合成的钙钛矿颗粒大小均一稳定,钙钛矿由粒状晶粒堆积而成,900 ℃合成的钙钛矿发生烧结,降低了催化剂的比表面积[15]。因此,选取800 ℃为适宜的焙烧温度。

a t=700 ℃

b t=800 ℃

c t=900 ℃图8 不同焙烧温度合成的CLM-0.5SEM照片
2.5 比表面积(BET)分析
CaMnO3和CLM-0.5的N2吸附-脱附曲线图见图9。
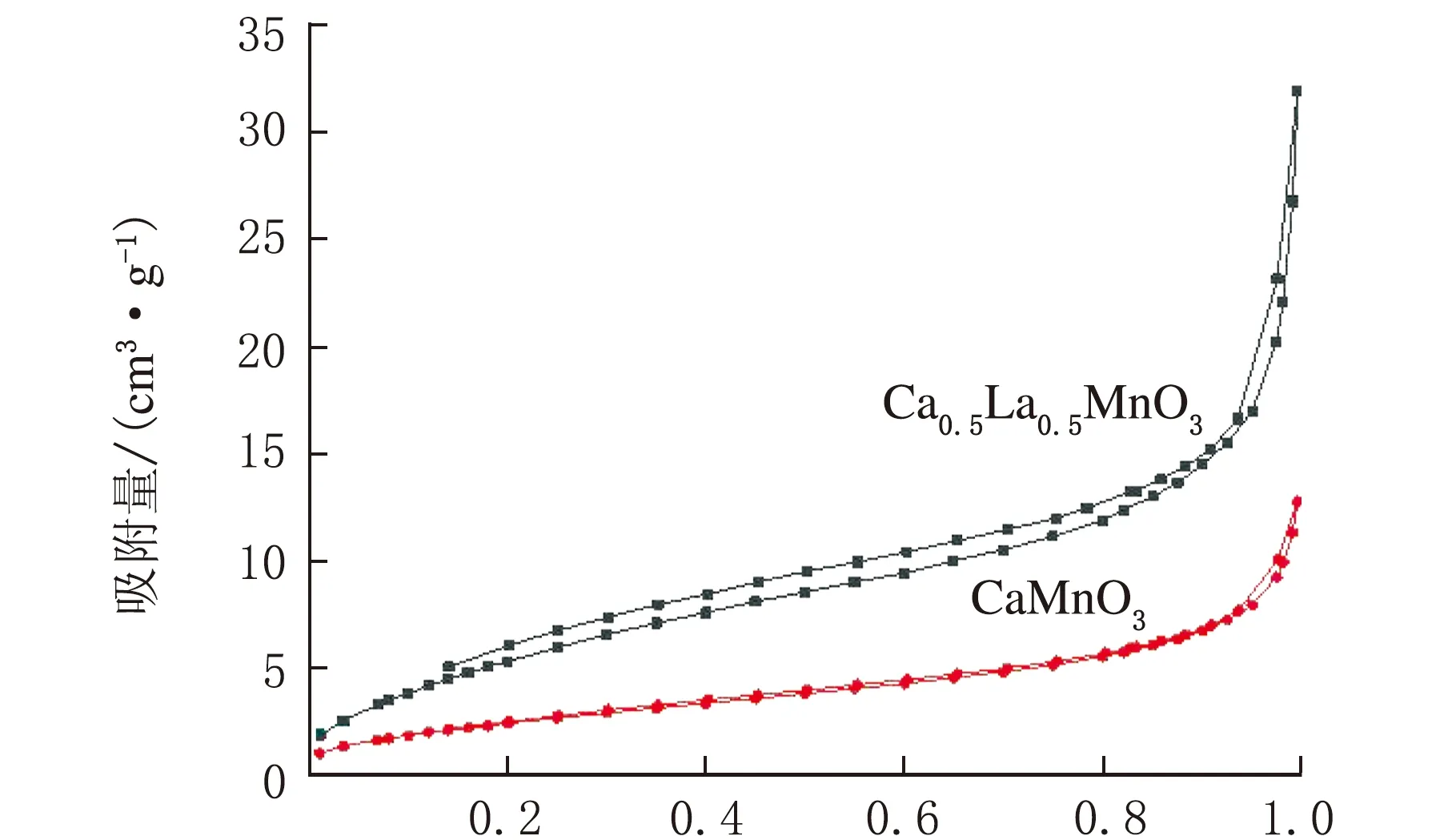
p/p0图9 CaMnO3和CLM-0.5的N2吸附-脱附曲线
由图9可知,相对压力较低时,样品所发生的吸附主要是以单分子层吸附为主,随着压力逐渐升高,由单分子层吸附转为多分子吸附,压力较高时,为多分子层吸附,最后出现毛细凝聚现象,此时,吸附曲线中可见一个突跃。由此可知,随着孔径逐渐提升,压力越高,毛细凝聚现象越易发生。CaMnO3样品和CLM-0.5样品吸附等温线均为Ⅳ型吸附等温线,钙钛矿的N2吸附-脱附曲线不相重合形成了滞后环,表明样品中存在介孔结构。
由BET分析得到的CaMnO3样品和CLM-0.5样品的结构参数见表1。

表1 CaMnO3样品和CLM-0.5样品孔结构参数
由表1可知,CaMnO3和CLM-0.5样品孔径分布为介孔结构。CLM-0.5样品的比表面积比CaMnO3样品增大了1.4倍。因此,通过掺杂La可提高锰酸钙的比表面积[16]。
2.6 木质素催化热解性能评价
木质素在不同钙钛矿作用下经热解反应得到的气、液、固三相产物分布见图10。
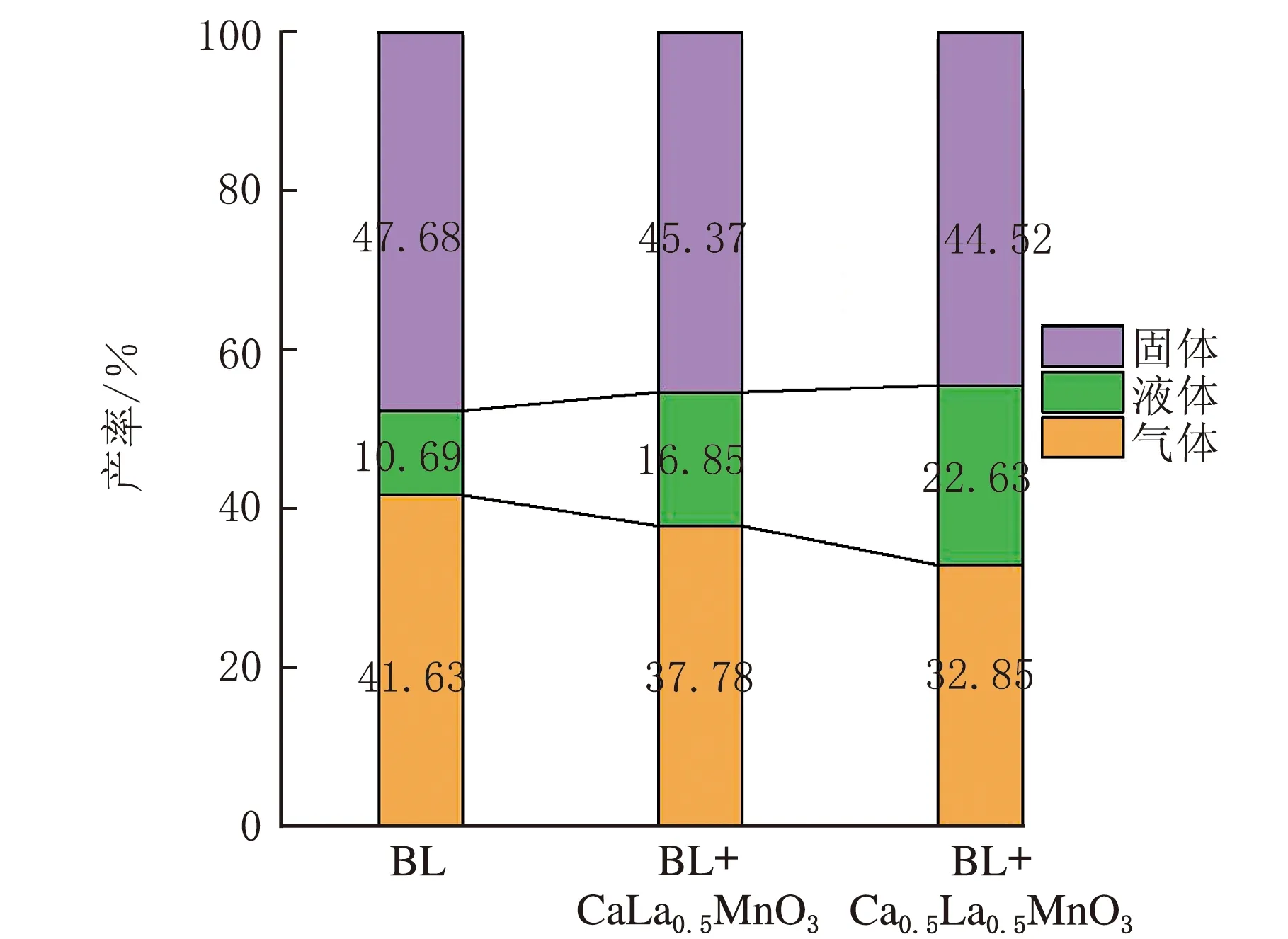
催化剂图10 不同催化剂作用下木质素热解气、液、固三相产物收率
由图10可知,未添加钙钛矿的纯木质素热解液体产物收率较低,木质素主要转化为CO、CO2、CH4和焦炭;木质素在CaMnO3催化剂作用下热解时液体产物产率提高至16.85%,并抑制气体产物的生成;由于La的掺杂使CLM-0.5催化剂的比表面积增大,增加了催化剂与木质素的接触面积,增强了固-固催化作用,同时由于La掺杂,将抑制木质素催化热解过程中脱侧链、脱羧和脱羰反应的发生,进而降低气体产物的收率,其气相产物收率约降低8%,对应固体焦炭产物的收率基本保持不变,因而,更多的裂解产物进入到液相产物中,对应液体产物收率最大,可达22.63%[17]。
BL在未添加催化剂与添加催化剂条件下热解的液体产物的组成分析见表2,液相产物各种物质的选择性分析见图11。
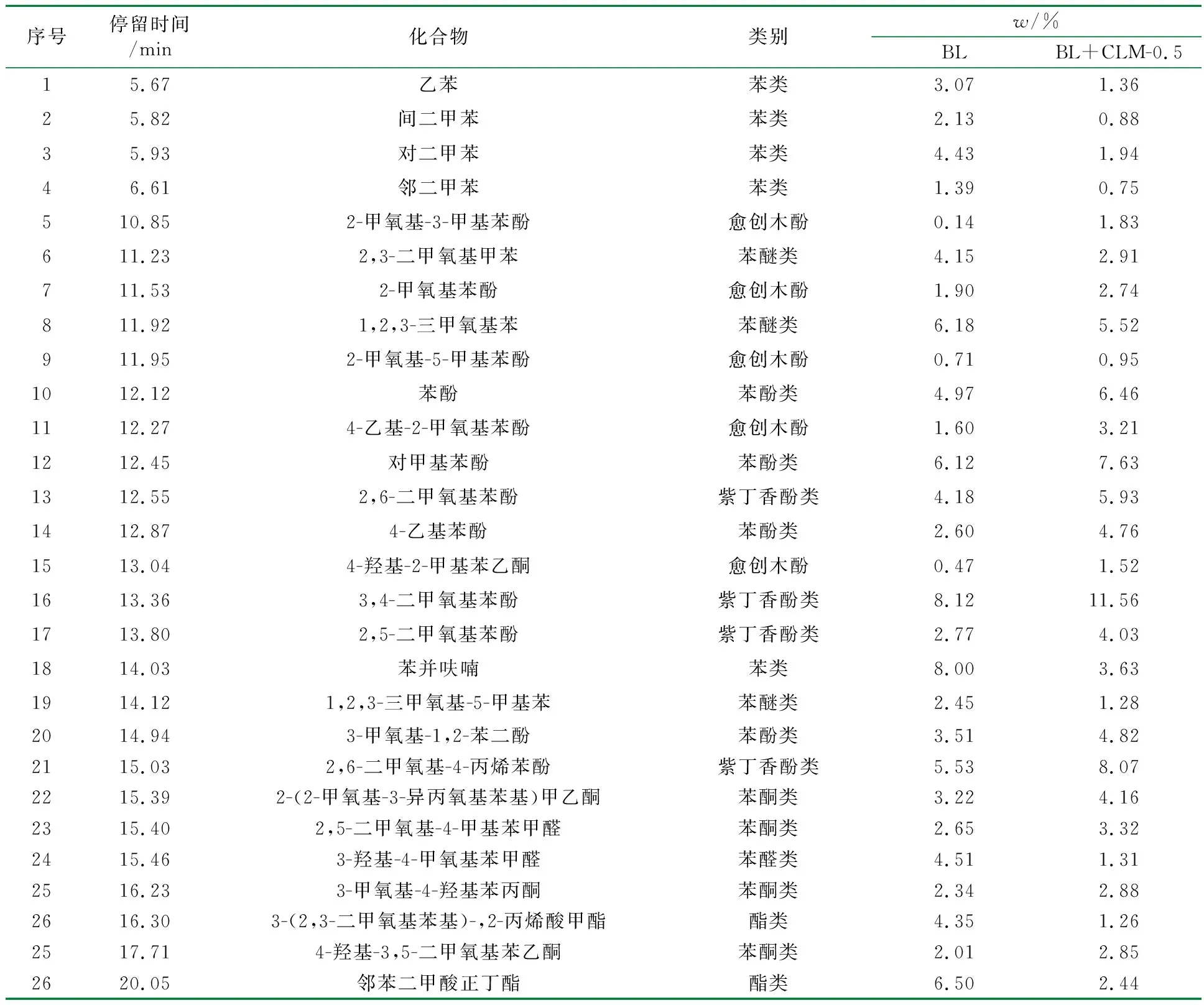
表2 GC/MS测定液体产品中芳香族化合物的含量
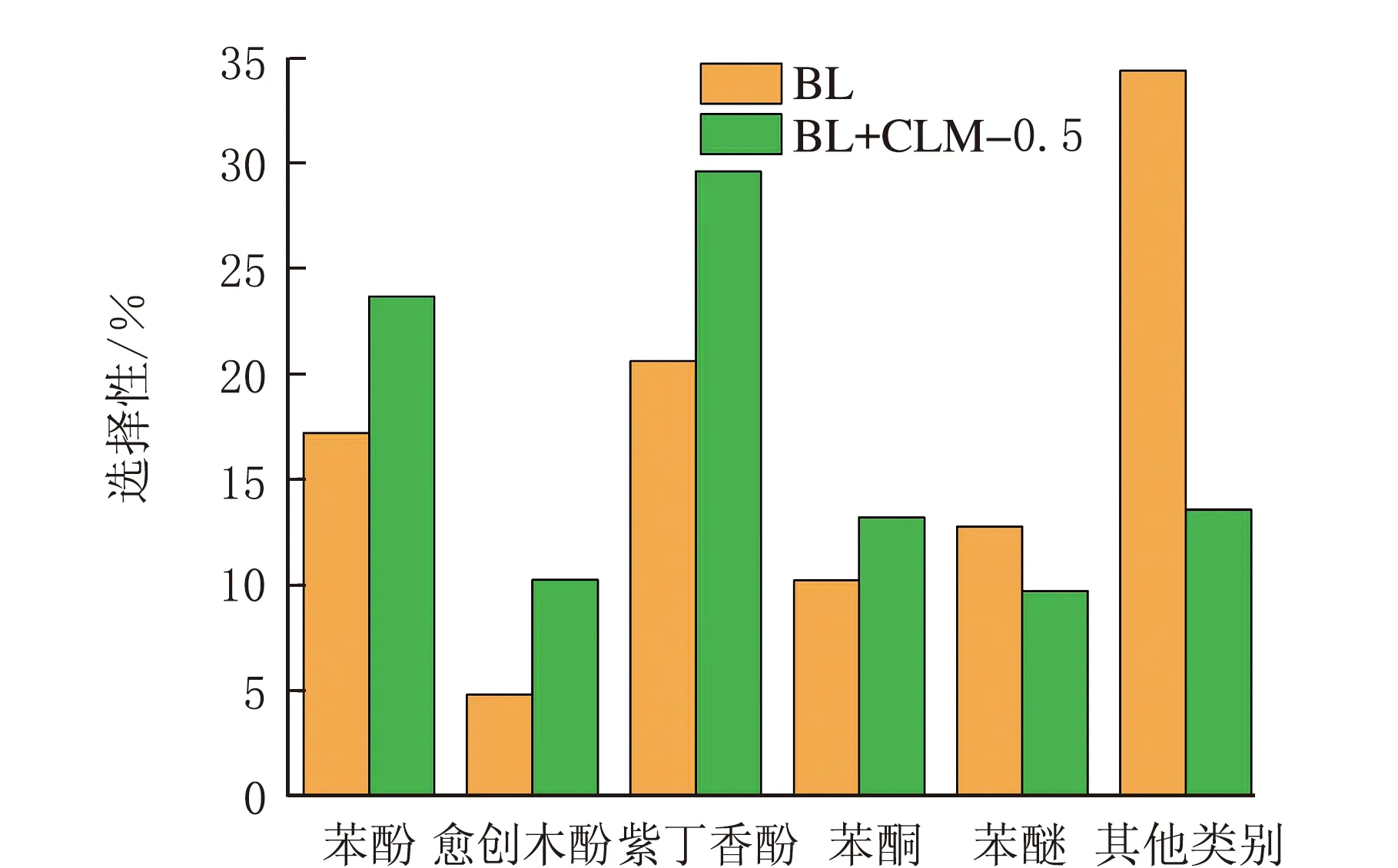
液相产物图11 液相产物各物质的选择性
2.7 催化剂的再生性能分析
催化剂的再生性能分析见图12。
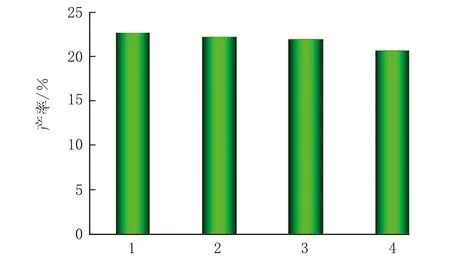
次数a 催化剂4次循环再生热解木质素液体产物收率
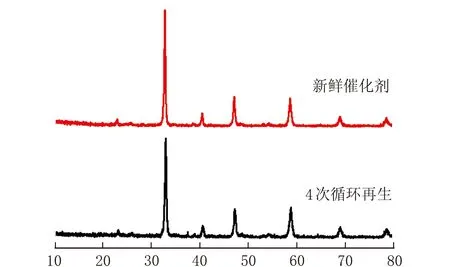
2θ/(°)b 新鲜催化剂与4次循环再生后催化剂XRD谱图

c 4次循环再生后催化剂SEM照片图12 催化剂再生表征分析
木质素催化热解后,钙钛矿表面包覆一层焦炭,通过氧化再生后可以实现其循环使用。由图12a可知,随着反应次数的增加,液体产物收率基本保持不变,说明CLM-0.5具有优异的再生性能;由图12b可知,经4次循环再生得到的CLM-0.5催化剂存在尖锐的特征峰,说明再生后的催化剂仍具有良好的结晶性,但相对于新鲜的催化剂特征峰变弱;由图12c可知,经4次热解再生得到的CLM-0.5催化剂颗粒粒径增大,并伴随烧结现象的出现,因此CLM-0.5再生过程中抑制烧结是今后研究的重点工作。
3 结 论
(1)溶胶凝-胶法合成镧掺杂的锰酸钙钛矿的适宜工艺参数为掺杂量x=0.5,前驱体pH=8,焙烧温度800 ℃,空气气氛下焙烧6 h,此时样品呈立方晶相,疏松多孔结构,其比表面积为22.65 m2/g;
(2)La掺杂有利于抑制木质素催化热解过程中脱侧链、脱羧和脱羰反应的发生,降低气体产物的收率,在固体焦炭产物收率基本保持不变的情况下,可以得到更多的液体产物,其收率为22.63%,产物中酚类化合物选择性提高21%;
(3)CLM-0.5催化热解木质素经氧化再生后形貌和晶相基本保持不变,说明催化剂具有良好的稳定性,经4次热解-再生后,木质素液相产物收率仍可达20%,说明再生后CLM-0.5具有较高的催化活性。
猜你喜欢
造纸信息(2022年8期)2022-11-10
物理学报(2022年1期)2022-01-19
轮胎工业(2021年4期)2021-12-25
燃烧科学与技术(2021年5期)2021-10-28
中国临床医学(2021年2期)2021-05-17
陶瓷学报(2020年6期)2021-01-26
陶瓷学报(2020年2期)2020-10-27
陶瓷学报(2020年2期)2020-10-27
新能源进展(2020年1期)2020-03-09
中国造纸(2019年6期)2019-09-10