
新型抗前列腺癌药物ODM-201的研究进展
2021-07-26 08:11:54黄晨超包旗旗张荣玉周金明
药学研究 2021年6期
黄晨超,包旗旗,张荣玉,周金明
(浙江师范大学化学与生命科学学院&现代制药创新研究中心,浙江 金华 321004)
前列腺癌是最常见的男性泌尿系统恶性肿瘤,病情发展缓慢,早期可以治愈,但是症状不明显,不易发现。晚期以保守治疗,延长患者生存期,维持生活质量为主。研究表明,2018年全球范围内,男性恶性肿瘤中前列腺癌发病率第二[1],有130万新发病例和36万死亡病例。我国前列腺癌的发病率相对较低,但受各方条件影响约束,大部分中国患者就诊时即处于晚期,且近十年来发病率也是逐年升高。
早在1941年,人们就发现通过手术去势或者使用雌激素治疗可以缓解晚期前列腺癌的症状。雄激素信号通路在前列腺癌的发生和发展中起到了重要作用[2],雄激素受体(androgen receptor,AR)是治疗前列腺癌最重要的靶点。80%以上的前列腺癌都是雄激素依赖性的,因而雄激素剥夺疗法(ADT)已成为前列腺癌男性患者全身性治疗的基础,在诊断后的第一年中,有44.8%的患者接受ADT,而在74岁以上且患有高级组织学指标诊断为T1、T2肿瘤的男性中,或患有T3、T4肿瘤的,这一比例增加到48.7%[3-7]。在病情前期,可以通过这种疗法有效控制病情。然而最终大部分患者的病情在经过中位时间18~24个月的缓和后,都会发展到受一种或多种抵抗机制驱动的去势抵抗性阶段[8-10],被称为去势抵抗性前列腺癌(CRPC)。
1 去势抵抗性前列腺癌及其治疗手段
根据2016年欧洲泌尿外科协会指南的标准[11],CRPC确诊需要同时满足两个条件:①血清睾酮达到去势水平(<1.7 mmol·L-1);②间隔一周,前列腺特异性抗原(prostate specific antigen,PSA)水平连续3次上调,且对较于最低值,升高超过50%,PSA>2 ng·mL-1,或者骨扫描发现2个及以上的新病灶或符合实体肿瘤评价标准的软组织病灶增大。
去势抵抗性前列腺癌发生机制最为常见的有3类。一是基因扩增[12]和过表达[13-15]导致AR的表达水平升高,对于肾上腺素来源与前列腺癌细胞自身合成的少量雄激素异常敏感[16-17]。二是基因突变产生AR突变体及可变剪接体。部分突变使得AR的激活不再局限于结合雄激素[12],失去了特异选择性。雌激素、糖皮质激素和孕激素等都可以与AR结合,促进前列腺癌细胞的生长;另一部分突变体可以使拮抗剂转化为激动剂[18],第一代抗雄激素药物尼鲁米特和氟他胺与T878A和H875Y结合后,反而使AR转为激动构象,AR信号通路被激活。另外一些AR可变剪接体由于缺少配体结合域,不需要结合雄激素就可以进入核内,发挥AR的调控功能,因而促进了去势抵抗的形成[19-21]。三是在AR信号通路上绕开AR进行信号传递或者完全不依赖于AR信号通路激活下游信号。糖皮质激素受体、黄体酮受体等同属于甾体核受体,在DNA结合域上和AR具有一定的相似性。这些受体蛋白的上调,可以补偿性激活AR下游信号通路,调控AR靶基因的表达[22-25]。除了AR信号通路外,Stat3信号通路的激活[26]或抗凋亡蛋白Bcl-2的上调[27]等都可以使前列腺癌细胞在缺少雄激素的情况下生长、增殖。
根据这些研究,我们不难发现,即使病情发展为CRPC,AR仍然可以作为前列腺癌治疗的重要靶点[28]。目前,CRPC的治疗药物主要有化疗药物,免疫抑制剂,放射剂,雄激素合成抑制剂和雄激素受体抑制剂等[29-32]。在用药阶段,需要综合考虑不同患者群体最适合的新药,CRPC患者的最佳给药时间和给药顺序,评估和确定新老药物联合使用的最佳组合,克服药物的耐药性等等问题。因此不断地研发新药,根据病人的具体情况联用用药,尽量延长病人的生存期,减轻痛苦是前列腺癌治疗不变的主题。目前正在研发的CRPC药物中主要方向是CYP17A1抑制剂及AR拮抗剂。
2 ODM-201的研究进展
由于AR信号通路对前列腺癌的重要意义,新型AR拮抗剂研发已成为抗前列腺癌药物研究的热点。二代AR拮抗剂相较于上一代,克服了一代拮抗剂引起的突变耐药性(如T878A会引起氟他胺耐药,而W742C会引起比卡鲁胺耐药),降低了不良反应的等级和发生频率,同时对AR的亲和性也有明显的提升。ODM-201(Darolutamide,BAY-1841788,NUBEQA)是拜耳与Orion联合推出的抗前列腺癌新药,是最新(2019年)通过美国食品药品监督管理局(FDA)批准的二代AR拮抗剂,用于非转移性去势抵抗性前列腺癌(non-metastatic castration resistant prostate cancer,nmCRPC)的治疗[33]。它与目前已有的AR拮抗剂在结构上有着明显的不同(见图1),在结合AR的亲和力以及抗肿瘤活性等方面都有不俗的表现。
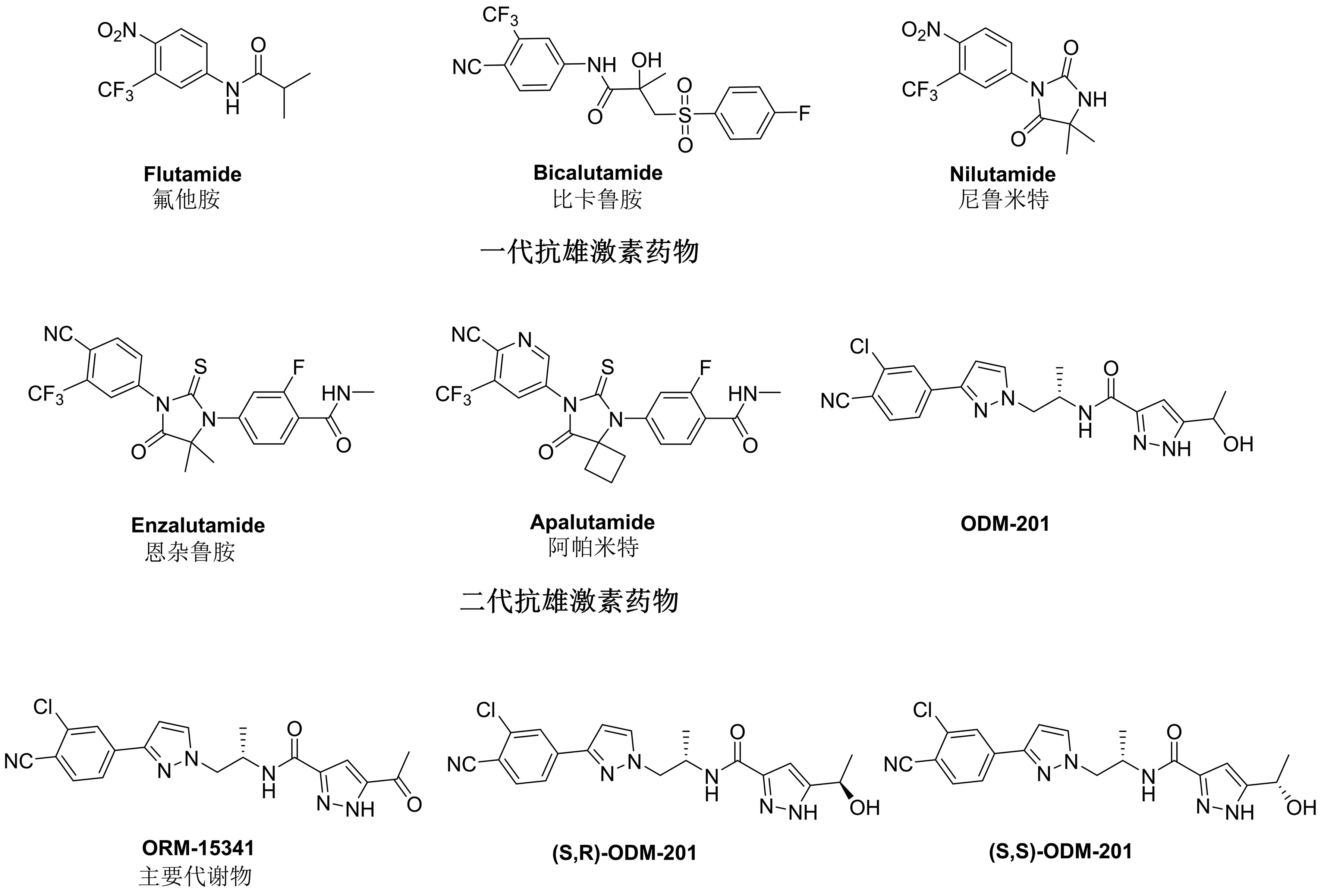
图1 一代、二代抗雄激素药物及ODM-201与其主要代谢物的结构
2.1 ODM-201结构及作用机理 ODM-201因其独特的化学结构而对AR具有很强的亲和力和拮抗活性,从而抑制了AR的功能和前列腺癌细胞的生长[34]。ODM-201化学名称为N-((S)-2-(3-(3-氯-4-氰基苯基)-1H-吡唑-1-基)-1-甲基乙基)-5-(1-羟基乙基)-1H-吡唑-甲酰胺。其中吡唑环上羟乙基具有两种构象,ODM-201为消旋化合物,同时具有(S,R)和(S,S)两种构象。体外和体内实验均证实了化合物是通过主要的循环代谢物ORM-15341实现这两个非对映异构体的快速转换(见图1)[34-35]。因此,两种异构体的拮抗作用基本相同,在合成时不需要额外选择手性合成。此外,尽管在高剂量下非对映异构体比例保持不变,但观察到的两种非对映异构体的药动学参数(PK parameter)略有不同,(S,R)构象的ODM-201可以更快地被代谢。
AR存在于细胞质内时与热休克蛋白结合,当雄激素与AR LBD域上的配体结合口袋结合后,诱导AR转变为激活构象,释放热休克蛋白形成二聚体。AR二聚体被转运进入细胞核内,与DNA上的特异应答元件作用,促进AR下游信号传递,从而促进细胞的生长与增殖。二代AR拮抗剂凭借与AR的高亲和性,竞争性地结合AR且使其不能被激活,进而阻断雄激素诱导的AR核易位。ODM-201的Ki值和IC50值分别为11和26 nmol·L-1,相较于恩杂鲁胺(86、219 nmol·L-1)和阿帕米特(93、200 nmol·L-1),具有明显的优势,同时其具有更加良好的体外抗细胞增殖活性及体内抗肿瘤活性[34]。2020年,拜耳针对ODM-201的作用机理进行的研究认为,ODM-201凭借对AR的高亲和力耗尽了AR基因调节区域,并且阻断了AR驱动的转录信号传导。根据评估H3K27ac、H3K4me1、FOXA1和BRD4的结合情况,推断增强子和超级增强子的激活在染色质水平被阻断,从而降低了几种重要的前列腺癌细胞增殖的下游途径,例如AR信号,脂肪酸代谢,未折叠的蛋白质反应以及PI3K/AKT/MTOR途径[36]。
除此之外,ODM-201较为独特的一点是,针对恩杂鲁胺和阿帕米特耐药的单突变体AR(F877L),恩杂鲁胺耐药的双突变体AR(H875Y/T878A)、AR(F877L/T878A)以及比卡鲁胺和氢化氟他胺耐药的AR(W742L)和 AR(T878A)都具有完全的拮抗作用。同时还有研究表明ODM-201显著抑制以前未报道过的比卡鲁胺和氟他胺耐药的突变体AR(T878G)的转录活性[34,37-38]。另外值得注意的是,ODM-201是唯一结合T878G突变体口袋却没有在高浓度下部分激活该AR突变体的药物[38]。对于这些突变体的抑制证明了ODM-201具有克服临床治疗中耐药性的潜力。
2.2 ODM-201的临床试验 ODM-201的ARADES(NCT01317641)和ARAFOR(NCT01784757)Ⅰ/Ⅱ期临床试验先后就ODM-201的长期服用安全性,耐受性和抗肿瘤活性进行了研究[39-40],结果均表明CRPC患者对于ODM-201的耐受度高,不良反应等级低,大多数不良事件为1-2级,并且不同剂量水平之间的不良事件特征没有差异。最常出现的不良反应(adverse events,ADs),如疲劳,腹泻,食欲下降,恶心等都被认为与疾病本身有关,而非药物引起。之前已有小鼠实验表明,口服 7 d后,ODM-201的血脑渗透率为1.9%~3.9%,而其他二代抗雄激素药物恩杂鲁胺和阿帕米特分别是27%和62%[34]。两次临床试验中均证实了这一点,ODM-201所诱发的如癫痫等中枢神经方面的副反应几乎可以忽略不计。ODM-201抗肿瘤活性强,在所有剂量和所有治疗分层亚组中均观察到PSA降低至少50%[39]。在总体人群中没有记录到明显的剂量依赖性活性,但是每天1 400 mg的剂量在先前未接受过化学疗法或CYP17抑制剂治疗的患者中产生的PSA响应率(PSA降低50%及以上的比率)最高,接受过CYP17抑制剂的患者最低[39]。这些数据与恩杂鲁胺和CYP17抑制剂相继治疗去势抵抗性前列腺癌中的交叉耐药性报道相符。试验结果还表明片剂和胶囊具有相似的药代动力学。但进食状态下,吸收率比禁食高两倍,表明与食物一起服用的片剂可以减轻患者的用药负担[40]。但是在2017年,日本国立癌症研究中心东医院就黄种人补充了ODM-201 Ⅰ期用药数据[41],由于试验条件控制不严,观察到的PSA响应率不算乐观,9名患者中仅1名在第12周PSA下降了50%以上。
ODM-201的ARAMIS Ⅲ期(NCT02200614)针对目前正在接受雄激素剥夺疗法治疗且疾病转移风险高的1 509名nmCRPC患者,评估了ODM-201的安全性和有效性[42-43]。根据试验结果,ODM-201可以显著延长中位无转移生存期。相较于安慰组,ODM-201组患者的中位无转移生存期延长了22个月[42]。ODM-201组和安慰剂组的3年总生存率分别为83%和77%。ODM-201组患者死亡或转移风险较安慰剂组显著降低59%[43]。最新公布的95名患者参与的日本亚组临床数据虽然在主要终点上未达到目标,但其余数据趋势均与总体保持一致,耐受性良好[44]。
2.3 ODM-201耐药的潜在机制 目前并没有明确针对ODM-201的耐药性研究报告,但2020年,Morsy等[45]研究发现,ODM-201可以在前列腺癌的体外模型中诱导AKR1C3(醛糖酮还原酶1C3)表达,并且对AKR1C3介导的耐药性敏感。AKR1C3参与内源性雄激素的合成,在前列腺癌细胞中高度表达会产生去势抵抗。研究人员用ODM-201处理AKR1C3不表达,正常表达和过表达的3种细胞系,得到IC50值与AKR1C3表达水平呈现正相关的结果。而耐药细胞经过高选择性的AKR1C3抑制剂KV-49g处理后,又重新对ODM-201恢复活性。由此推断AKR1C3过表达很有可能是ODM-201的潜在耐药机制,可以通过联合用药克服。
AKR1C3过表达已经被证明是恩杂鲁胺/阿比特龙耐药性的关键机制[46-48]。此外,目前包括ODM-201在内的二代AR拮抗剂都无法靶向AR-V7可变剪接体,因此临床上认为AR-V7在恩杂鲁胺耐药机制中也起着重要作用。有研究表明AKR1C3可以通过泛素-蛋白酶体系统调节和稳定全长AR和AR-V7可变剪接体[46]。这表明AKR1C3/AR-V7轴在恩杂鲁胺和阿比特龙的耐药中起关键作用。对于恩杂鲁胺/阿比特龙耐药细胞,ODM-201具有交叉耐药性。但是通过敲除或者靶向耐药细胞中的AKR1C3蛋白可以显著下调全长AR、AR-V7、c-Myc的表达,使耐药细胞对ODM-201重新敏感。然而敲除或者靶向AR-V7可变剪切体只会下调c-Myc,而不影响AKR1C3的表达[49],癌细胞对于ODM-201仍然具有耐药性。
2.4 ODM-201的合成工艺研究 在ODM-201通过了FDA批准后,整个药物和药品开发计划都将由Orion执行。Orion公司优化了重要的工艺开发提高了收率,通过结晶进行了流水线分离,并尽量避免了不经济的试剂,但根据数项披露的专利,可知ODM-201的合成途径仍未改变[50]。Orion公司设计和开发的ODM-201的最新合成路线如图2所示。
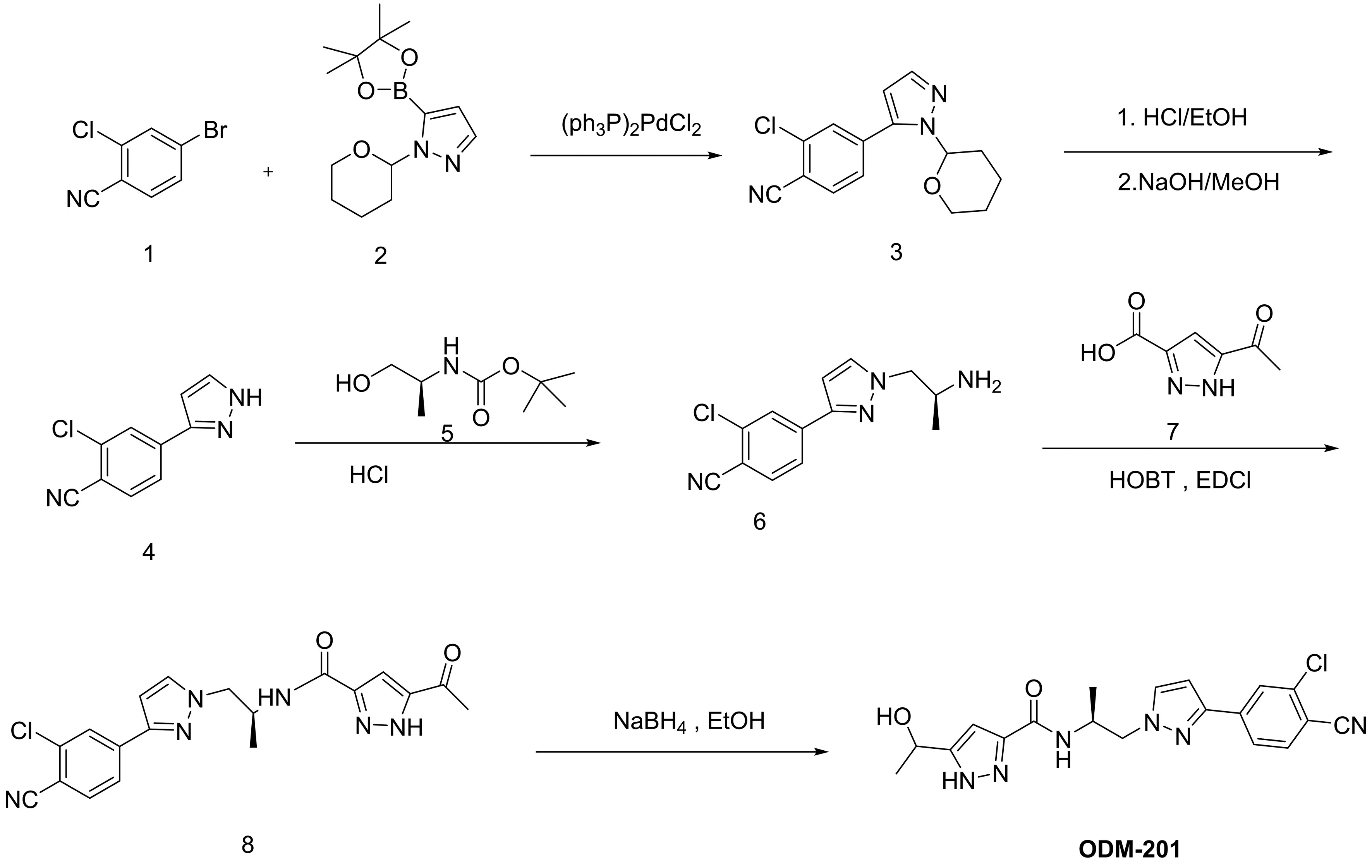
图2 Orion公司公布的合成路线
ODM-201是非对映异构体的混合物。两种单一构象的化合物可以根据专利WO 2016/1230530 A1进行合成[51],如图3A所示。其中关键手性中间体10、11可分别通过KRED酶(酮还原酶)催化得到,或者通过化合物12和手性化合物13或14在催化剂下分别制备得到[51]。
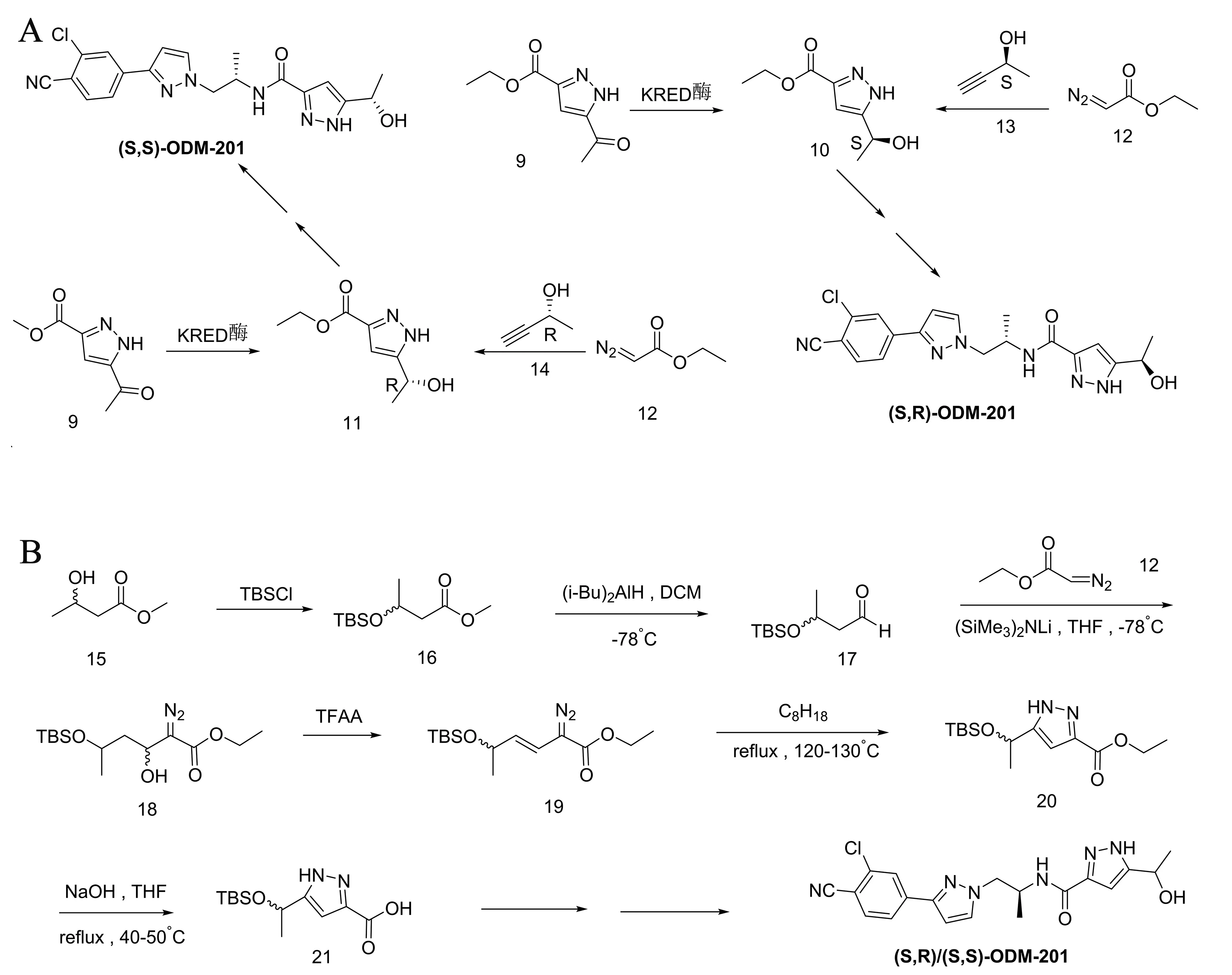
图3 ODM-201手性合成工艺
上述手性合成的方法中,酶还原不适合应用于工业生产。后者反应收率极低,仅为0.026%。因此2017年,Pan等[52]利用分子内1,3-偶极环加成反应开发了一种高效合成手性ODM-201的方法。将化合物20手性吡唑环羧酸乙酯的产率大大提高,但由于反应路线略显冗长,8步反应总收率为0.4%~0.7%。目前更为简便,合理,适用于工业生产ODM-201的方法还有待开发。
2.5 基于ODM-201的结构优化 到目前为止,针对ODM-201结构优化方面的研究还很少。2019年,四川大学的Yu等[53]针对ODM-201的结构进行优化,以期获得抗肿瘤活性更好的抗雄激素药物前体,得到了一批针对野生型AR和AR突变体的双功能(下调剂,抑制剂)ODM-201类似物,同时选出了能够克服CRPC耐药性,有效且口服可用的先导化合物4k。
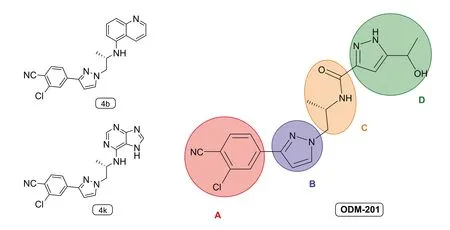
图4 ODM-201及其类似物4k、4b的化学结构
将ODM-201结构简单分为4个部分,A、B、D环和C链,主要针对D环和C链进行了改造。根据表1中数据显示,ODM-201及ORM-15341在VCaP细胞系中浓度增大到一定程度抑制作用反而减弱。用不同取代基的苯基或烷基取代D环上的吡唑环部分,所合成的化合物在VCaP细胞系变为浓度依赖性抑制,说明针对D环部分的改造是有意义的。结构上应当保留芳香环,且芳香环上取代基的影响巨大。苯环间位取代相比对位活性更强。去掉C链上的羰基,并在D环处引入了各种苯环,芳香杂环,芳香族二环,绝大部分化合物表现出良好至优异的活性,同时部分化合物对于表达AR-V7的22Rv1细胞系表现出了抑制活性,IC50值降低至亚微摩尔范围。证明羰基可以消除,芳香环对于活性起到正向作用,且杂环活性大部分高于苯环,氮的个数和取代位点对于活性影响极大,但是具体的影响机理从目前的数据中还无法判断。
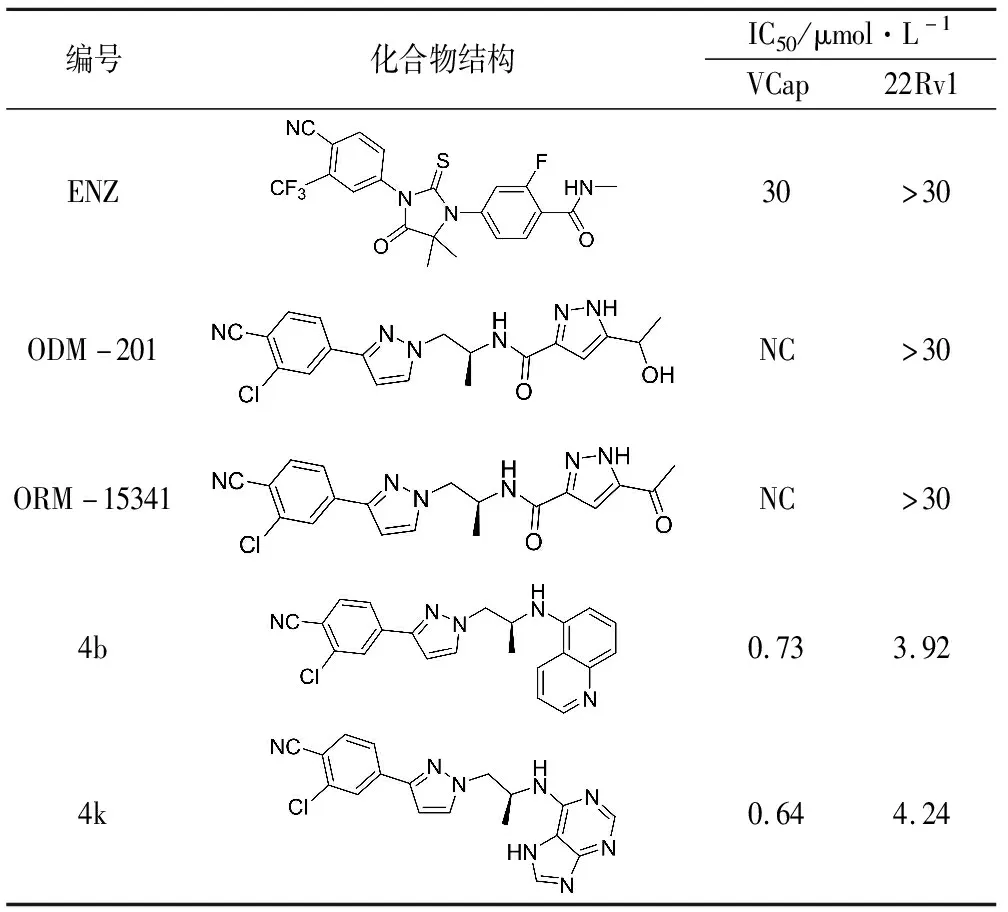
表1 部分化合物的体外抗细胞增殖活性[53]
最终发现,连接喹啉(4b)和嘌呤(4k)基团的化合物对AR过表达的VCaP细胞和AR-V7阳性22Rv1细胞的抗增殖活性优于ODM-201和恩杂鲁胺。筛选包括4b、4k在内活性尚可的化合物进入下一步研究,发现这些化合物基本上都能在mRNA水平下调AR的表达,其中4b和4k仍然是下调效果最佳的。4k和4b对于全长AR、AR-V7、AR-F876L突变体有抑制和下调作用,4k在体内半衰期比较短,但是有更高的最大血浆浓度(2 648.88 ng·mL-1)和全身暴露(AUC0~t值为7 280.76 ·h·mL-1)[53],因此用于动物实验,结果表明4k的抗肿瘤活性优于恩杂鲁胺,在治疗期间未观察到明显的体重减轻。
3 总结与展望
ODM-201作为一个全新结构的AR拮抗剂,对于多种AR突变体具有抑制作用,具有在临床治疗中克服耐药的潜力,能够显著抑制肿瘤增长,不会透过血脑屏障,产生中枢神经方面的不良反应。临床试验证明它能够显著延长nmCRPC患者的中位无转移生存期,目前还未产生耐药,建议在病情发展早期ADT联合ODM-201进行治疗。虽然ODM-201已经通过了FDA批准,由公司进行生产和销售,但是其工业化生产的合成路线仍然值得进一步优化。由于ODM-201结构独特,目前也没有大量研究对其进行优化,因此构效关系不够明确,只能确定维持C链的S构象,丙胺基对于药物活性有利,D环上氮杂环优于苯环,氮的个数和位置影响极大,但具体如何影响尚不明确。
目前ODM-201上市最大的困难应该是与恩杂鲁胺竞争市场份额。同时随着药物上市后更多的临床数据,可能会出现延迟和罕见的不良反应,需要对ODM-201研究的进一步深入以及结构优化以期得到效果更好的抗雄激素药物。
猜你喜欢
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
海南医学(2016年8期)2016-06-08 05:43:00
中国社区医师(2015年14期)2015-12-24 00:37:31
生殖医学杂志(2015年11期)2015-02-28 16:32:16
山东医药(2015年40期)2015-02-28 14:28:45
中华皮肤科杂志(2014年4期)2014-12-19 12:55:43
今日畜牧兽医(2014年1期)2014-10-09 05:45:56
当代畜禽养殖业(2014年7期)2014-02-27 07:59:14
食品科学(2013年23期)2013-03-11 18:30:11
