
正交试验优化白花蛇舌草4种五环三萜类成分提取工艺
2021-07-25 00:31林艾和刘海鹏张艳娇黄宽李蓉范源
中国中医药信息杂志 2021年6期
林艾和,刘海鹏,张艳娇,黄宽,李蓉,范源,
正交试验优化白花蛇舌草4种五环三萜类成分提取工艺
林艾和1,刘海鹏2,张艳娇1,黄宽1,李蓉1,范源1,2
1.云南中医药大学,云南 昆明 650500;2.云南中医药大学第二附属医院,云南 昆明 650216
优选白花蛇舌草五环三萜类成分山楂酸(MA)、科罗索酸(CA)、齐墩果酸(OA)、熊果酸(UA)的最佳提取工艺。采用HPLC建立MA、CA、OA、UA含量测定方法,以MA、CA、OA、UA含量为评价指标,通过单因素试验和正交试验,考察乙醇体积分数、提取时间、提取次数对提取效果的影响。MA、CA、OA、UA色谱峰分离度良好,分别在0.568~3.408 μg(2=0.999 0)、0.582~3.492 μg(2=0.999 0)、0.488~2.928 μg(2=0.999 9)、0.720~4.320 μg(2=0.999 2)范围内呈良好的线性关系,平均加样回收率分别为99.09%(RSD=1.57%)、99.76%(RSD=1.78%)、100.49%(RSD=1.66%)、99.52%(RSD=1.75%)。最佳提取工艺条件为:采用95%乙醇提取70 min,提取3次。本研究优选的提取工艺合理、稳定可行,可用于白花蛇舌草中MA、CA、OA、UA的提取。
白花蛇舌草;正交试验;提取工艺;山楂酸;科罗索酸;齐墩果酸;熊果酸
白花蛇舌草为茜草科植物白花蛇舌草Willd的干燥全草[1],广泛分布于我国浙江、江苏、广东、江西、云南等地[2],其性寒,味苦、甘,归胃、大肠、小肠经,具有清热解毒、活血止痛、利尿消肿等功效,临床用于治疗湿热黄疸、小便不利、咽喉肿痛、疮疖肿痛、阑尾炎、毒蛇咬伤、盆腔炎、泌尿系感染等。白花蛇舌草主要含有多糖类、三萜类、有机酸类、黄酮类、蒽醌类、烷烃类、甾醇类、环烯醚苷萜类、生物碱等化学成分[3-5]。其中,三萜类化合物具有抗菌、消炎、抗癌、抗突变、抗氧化等药理作用[6-9]。白花蛇舌草中三萜类化合物多为乌苏烷型和齐墩果烷型,其中2组同分异构体化合物熊果酸(ursolic acid,UA)和齐墩果酸(oleanolic acid,OA)、科罗索酸(corosoic acid,CA)和山楂酸(maslinic acid,MA)含量较高,结构和化学性质相似,色谱峰出峰时间接近,分离测定存在一定难度[10-14]。本研究建立白花蛇舌草中MA、CA、OA和UA的HPLC定量分析方法,在单因素试验基础上,采用正交试验优化其提取工艺,为白花蛇舌草的开发利用提供依据。
1 仪器与试药
YL-040S语路超声波清洗机,深圳市即洁超声科技有限公司;Agilent 1200高效液相色谱仪,DAD检测器,Acclaim C30色谱柱(2.1 mm×150 mm,3 μm),美国Agilent公司;DFY-600摇摆式高速万能粉碎机,永康市速锋工贸有限公司;D2KW-D电热恒温水浴锅,上海量壹科学仪器有限公司;T-1000型电子天平,上海浦春计量仪器有限公司;AB265-S十万分之一分析天平,Mettler-Tolido International Trade(Shanghai)Co.LTD;旋转蒸发仪,上海爱朗仪器有限公司。
白花蛇舌草(批号P20190912,安国市旭芳中药材经营有限公司),经云南中医药大学李国栋副教授鉴定为茜草科植物白花蛇舌草Willd的干燥全草。对照品MA(批号wkq18080306)、CA(批号wkq20042404)、OA(批号wkq18050212,)、UA(批号wkq20061211),纯度≥98%,四川省维克奇生物科技有限公司。甲醇、乙腈为色谱纯,其余试剂为分析纯。
2 方法与结果
2.1 指标成分测定方法
2.1.1 色谱条件
色谱柱:Acclaim C30色谱柱(2.1 mm×150 mm,3 μm);流动相:甲醇(B)-乙腈(C)-0.1%磷酸水溶液(D);梯度洗脱(0~10 min,75%B、12.5%C、12.5%D;10~20 min,70%B、15%C、15%D;20~25 min,75%B、12.5%C、12.5%D);流速:0.1 mL/min;检测波长:210 nm;柱温:20 ℃;进样量:10 μL。理论塔板数按MA峰计算应不低于5000,分离度均大于1.5。色谱图见图1。
2.1.2 混合对照品溶液制备
分别精密称取对照品MA 2.84 mg、CA 2.91 mg、OA 2.44 mg、UA 3.60 mg,置10 mL量瓶中,用甲醇配制成MA、CA、OA、UA浓度分别为0.284、0.291、0.244、0.360 mg/mL的混合对照品溶液,低温避光保存。
2.1.3 供试品溶液制备
取白花蛇舌草样品,粉碎,过40目筛,精密称定约1.0 g,置于圆底烧瓶中,进行回流提取,提取温度90 ℃,料液比为1∶20,设置不同乙醇体积分数、提取时间及提取次数,合并提取液,过滤,滤液于60 ℃减压回收溶剂至干,加50 mL水使混悬,用乙酸乙酯振摇萃取5次,每次50 mL,合并乙酸乙酯液,减压回收溶液至干,残渣加甲醇移至10 mL量瓶中,加甲醇稀释至刻度,摇匀,用0.45 μm微孔滤膜过滤,取续滤液,即得。

注:A.对照品;B.供试品;1. MA;2. CA;3. OA;4. UA
2.1.4 线性关系考察
取“2.1.2”项下混合对照品溶液,0.45 μm微孔滤膜过滤,依次精密吸取2、4、6、8、10、12 μL,按“2.1.1”项下色谱条件进样,记录色谱图。以对照品进样量(μg)为横坐标,峰面积为纵坐标,进行线性回归。MA回归方程为=457.13-168.66(2=0.999 0),线性范围为0.568~3.408 μg;CA回归方程为=1007.7-398.94(2=0.999 0),线性范围为0.582~3.492 μg;OA回归方程为=657.6-70.54(2=0.999 9),线性范围为0.488~2.928 μg;UA回归方程为=1774.9-353.47(2=0.999 2),线性范围为0.720~4.320 μg。
2.1.5 精密度考察
精密吸取同一混合对照品溶液10 μL,按“2.1.1”项下色谱条件连续进样6次,结果MA、CA、OA、UA的峰面积RSD分别为1.92%、1.95%、1.69%、1.65%,表明仪器精密度良好。
2.1.6 稳定性试验
按“2.1.3”项下方法制备供试品溶液,于室温放置0、2、4、6、8、10 h,按“2.1.1”项下色谱条件进样分析,结果MA、CA、OA、UA峰面积RSD分别为1.70%、1.63%、1.95%、0.61%,表明供试品溶液在10 h内稳定。
2.1.7 重复性试验
取同一批样品6份,按“2.1.3”项下方法制备供试品溶液,按“2.1.1”项下色谱条件进样分析,结果MA、CA、OA、UA含量RSD分别为1.28%、1.46%、1.24%、1.68%,表明该方法重复性良好。
2.1.8 加样回收率试验
精密称取已知含量的同一批样品(每1.0 g样品含MA 0.893 3 mg、CA 1.736 0 mg、OA 1.196 6 mg、UA 3.487 1 mg)粉末6份,每份精密称取1.0 g,按“2.1.3”项下方法制备供试品溶液,加入约等量MA、CA、OA、UA对照品,按“2.1.1”项下色谱条件进样测定,计算平均回收率,结果见表1。
表1 白花蛇舌草中MA、CA、OA、UA加样回收率试验
成分取样量 /g样品中含量/mg加入量 /mg测得量 /mg回收率 /%平均回收率/%RSD /% MA1.013 70.850 40.927 41.792 3100.82 99.091.57 1.002 70.923 20.922 11.802 8 97.70 1.033 60.893 30.892 51.785 5 99.98 1.028 10.879 40.910 41.792 4100.15 1.047 70.913 20.905 11.802 1 99.11 1.029 40.925 00.872 31.739 6 96.79 CA1.014 01.685 61.745 53.387 6 98.73 99.761.78 1.017 21.703 11.694 23.325 9 97.90 1.068 21.736 01.727 13.502 8101.15 1.023 21.724 31.680 63.346 3 98.28 1.053 41.690 81.703 33.476 0102.41 1.058 11.783 61.698 83.485 2100.08 OA1.005 21.208 41.170 62.451 3103.04100.491.66 1.017 81.170 51.209 12.380 5100.04 1.028 91.196 61.217 92.377 3 98.46 1.085 71.194 11.164 52.385 3101.13 1.047 41.238 21.185 92.400 6 99.03 1.022 61.214 91.230 12.475 7101.26 UA1.039 73.301 63.597 86.834 7 99.0699.521.75 1.003 73.472 83.329 56.801 9 99.99 1.033 53.487 13.502 16.890 2 98.58 1.069 13.610 93.593 76.991 6 97.04 1.029 63.387 23.370 16.771 2100.21 1.002 33.602 13.580 37.342 3102.23
2.2 单因素试验
采用加热回流法提取白花蛇舌草中MA、CA、OA、UA。按“2.1.3”项下方法制备供试品溶液,考察乙醇体积分数、提取时间及提取次数对指标成分含量(mg/g)的影响。指标成分含量(mg/g)=供试品峰面积×对照品浓度(mg/mL)÷对照品峰面积。
2.2.1 乙醇体积分数
固定提取时间为70 min、提取次数为1次,考察不同乙醇体积分数(65%、75%、85%、95%)对MA、CA、OA、UA提取效果的影响,结果见图2。随着乙醇体积分数增加,MA、CA、OA、UA含量逐渐升高。综合考虑,选择乙醇体积分数为95%。

图2 乙醇体积分数对白花蛇舌草MA、CA、OA、UA提取效果的影响
2.2.2 提取时间
固定乙醇体积分数为95%、提取次数为1次,考察不同提取时间(50、70、90、110、130 min)对MA、CA、OA、UA提取效果的影响,结果见图3。MA含量在提取50~70 min增加,70~130 min减少;CA含量在提取50~130 min逐渐增加;OA含量在提取50~70 min增加,70~130 min减少;UA含量在提取50~70 min增加,此后较70 min均减少。综合考虑,选择提取时间为70 min。
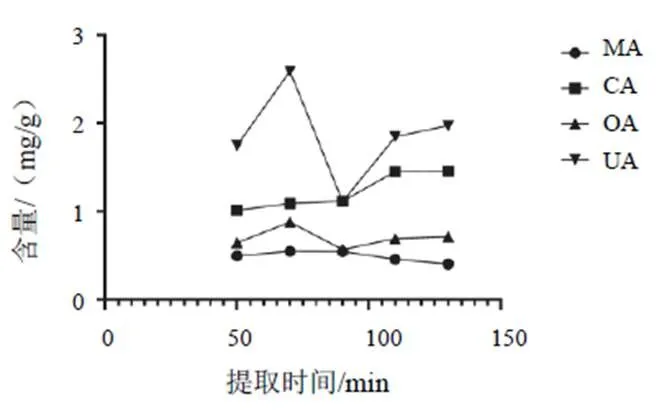
图3 提取时间对白花蛇舌草MA、CA、OA、UA提取效果的影响
2.2.3 提取次数
固定乙醇体积分数为95%、提取时间为70 min,考察不同提取次数(1、2、3、4次)对MA、CA、OA、UA提取效果的影响,结果见图4。MA、OA含量随提取次数增加而增大;CA、UA含量在提取3次时最高,提取4次时减少。综合考虑,选择提取次数为3次。

图4 提取次数对白花蛇舌草MA、CA、OA、UA提取效果的影响
2.3 正交试验
2.3.1 正交试验因素与水平
选取乙醇体积分数、提取时间、提取次数进行3因素3水平正交试验,考察各因素对MA、CA、OA、UA含量的影响,因素水平见表2。
表2 正交试验因素水平表
水平乙醇体积分数/%A提取时间/minB提取次数C 175 701 285 902 3951103
2.3.2 正交试验结果及方差分析
分别精密称取白花蛇舌草粉末1.0 g,按正交试验设计进行试验,按“2.1.3”项下方法制备供试品溶液,按“2.1.1”项下色谱条件进样测定,计算MA、CA、OA、UA含量,并进行方差分析,结果见表3~表7。
表3 白花蛇舌草中4种成分L9(33)正交试验结果(mg/g)
试验号ABCMACAOAUA 11110.5020.7280.7102.344 21220.4230.6700.6302.173 31330.3440.9900.7492.673 42120.6071.1930.9592.906 52230.5941.2430.8341.705 62310.2490.7110.4611.277 73130.9261.7031.2533.493 83210.5461.1180.5681.115 93320.4360.8130.5891.075 K1(MA)0.4230.6780.432 K1(CA)0.7961.2080.852 K1(OA)0.6960.9740.580 K1(UA)2.3972.9141.579 K2(MA)0.4830.5210.489 K2(CA)1.0491.0100.892 K2(OA)0.7510.6770.726 K2(UA)1.9631.6642.051 K3(MA)0.6360.3430.621 K3(CA)1.2110.8381.312 K3(OA)0.8030.6000.945 K3(UA)1.8941.6752.624 R(MA)0.2130.3350.189 R(CA)0.4150.3700.460 R(OA)0.1070.3740.365 R(UA)0.5031.2501.045
表4 MA含量正交试验结果方差分析
因素偏差平方和自由度F比F临界值显著性 A0.07221.1256.940 B0.16922.6416.940P<0.05 C0.05620.8756.940 误差0.1304
表5 CA含量正交试验结果方差分析
因素偏差平方和自由度F比F临界值显著性 A0.26321.27719.000 B0.20621.00019.000 C0.38921.88819.000 误差0.2102
表6 OA含量正交试验结果方差分析
因素偏差平方和自由度F比F临界值显著性 A0.0172 1.00019.000 B0.234213.76519.000P<0.05 C0.203211.94119.000P<0.05 误差0.0202
表7 UA含量正交试验结果方差分析
因素偏差平方和自由度F比F临界值显著性 A0.44521.00019.000 B3.09926.96419.000P<0.05 C1.64323.69219.000 误差0.4502
以MA含量为指标,3个因素对提取效果的影响为B>A>C;以CA含量为指标,3个因素对提取效果的影响为C>A>B;以OA含量为指标,3个因素对提取效果的影响为B>C>A;以UA含量为指标,3个因素对提取效果的影响为B>C>A。以A、C为误差列分别对MA含量进行方差分析,结果B因素对MA含量影响显著;以B为误差列对CA含量进行方差分析,结果A、B、C因素均对CA含量无显著影响;以A为误差列对OA含量进行方差分析,结果B、C因素对OA含量影响显著;以A为误差列对UA含量进行方差分析,结果B因素对UA含量影响显著。
提取工艺A3B1C3的提取效果最佳,在此条件下MA、CA、OA和UA含量分别为0.926、1.703、1.253、3.493 mg/g。综合考虑,最佳提取工艺组合为A3B1C3,即95%乙醇提取70 min,提取3次。
2.3.3 验证试验
平行称取样品粗粉3份,每份1.0 g,精密称定,按优选工艺进行验证,结果见表8。表明正交试验优选的工艺条件较好,合理可行。
表8 白花蛇舌草中4种成分提取工艺验证试验结果(mg/g)
编号MACA OA UA 10.9311.6691.2743.527 20.8951.6711.2163.412 30.9181.7321.2723.528 平均含量0.9151.6911.2543.489 RSD/%1.992.122.631.91
3 讨论
MA、CA、OA和UA均为白花蛇舌草中的五环三萜类化合物,MA与CA、OA与UA分别为同分异构体,结构相似,MA是OA的衍生物,CA是UA的衍生物。这2组同分异构体在中性流动相中可解离,常规色谱条件很难使其达到基线分离。本研究在多次预试验基础上,发现以甲醇、乙腈、磷酸共同作为流动相使用能很好提高其分离度,改善色谱峰的峰形,出峰时间适合。经反复试验,调整甲醇-乙腈-0.1%磷酸水溶液三者的比例,最终以甲醇(B)-乙腈(C)- 0.1%磷酸水溶液(D)为流动相梯度洗脱,2组同分异构体的分离度均大于1.5,达到了检测要求。进而以MA、CA、OA和UA含量为指标,在单因素试验基础上,采用正交试验对影响提取效果的乙醇体积分数、提取时间、提取次数进行分析,得到最佳提取工艺参数。
白花蛇舌草临床应用广泛,但现有的质量控制研究文献报道较少,其优化提取工艺多以UA为评价指标。OA是UA同分异构体,MA为OA衍生物,CA为UA衍生物且二者含量较高,仅以UA为检测指标存在局限性,难以较好优化其提取工艺。本试验以MA、CA、OA、UA为指标成分,指标选择更为合理,结果更准确,更适用于白花蛇舌草有效成分的提取工艺优化。
[1] 国家药典委员会.中华人民共和国药典:一部[M].北京:中国医药科技出版社,2010:附录23.
[2] 国家中医药管理局中华本草编委会.中华本草精选本[M].上海:上海科学技术出版社,1998:433.
[3] 李波.白花蛇舌草的化学成分和药理作用研究进展[J].天津药学, 2016,28(5):75-78.
[4] 邹香妮,闫珺.白花蛇舌草抗肿瘤化学成分及作用机制研究进展[J].化学工程师,2020,34(3):47-49,56.
[5] 杨新周,田孟华,杨子仙,等.白花蛇舌草中黄酮、多酚、多糖提取工艺及抗氧化研究[J].化学研究与应用,2019,31(1):22-30.
[6] WANG C, ZHOU X, WANG Y, et al. The antitumor constituents fromWilld[J]. Molecules,2017,22(12):2101.
[7] 刘枭,许云,徐银莹,等.白花蛇舌草治疗子宫肌瘤机制的网络药理学研究[J].中药新药与临床药理,2020,31(9):1070-1078.
[8] 王骁,范焕芳,李德辉,等.白花蛇舌草的抗癌作用研究进展[J].中国药房,2019,30(10):1428-1431.
[9] 车宏伟,杨海宁,谭晓川,等.白花蛇舌草不同提取部位抗肿瘤作用研究进展[J].亚太传统医药,2019,15(3):191-193.
[10] 吴燕.高效液相色谱法测定不同产地白花蛇舌草中齐墩果酸的含量[J].海峡药学,2016,28(5):58-59.
[11] 王欢,吴莹,黄嫣,等.HPLC-DAD同时测定白花蛇舌草中齐墩果酸和熊果酸的含量[J].中药与临床,2016,7(1):20-22,36.
[12] 李敏,齐红,于姗姗,等.市售白花蛇舌草正品及伪品中齐墩果酸和熊果酸的含量测定[J].中国药物经济学,2020,15(7):49-52.
[13] 杨新周,吴娜,邱晓雪.HPLC法测定云南不同产地白花蛇舌草中齐墩果酸和熊果酸含量[J].贵州农业科学,2019,47(10):115-118.
[14] 刘慧妍,黄馨慧,张艳海.HPLC法快速测定8种中药中齐墩果酸和熊果酸[J].中草药,2017,48(10):1998-2001.
Optimization of Extraction Technology of Four Pentacyclic Triterpenes fromby Orthogonal Experiment
LIN Aihe1, LIU Haipeng2, ZHANG Yanjiao1, HUANG Kuan1, LI Rong1, FAN Yuan1,2
To optimize the extraction technology of pentacyclic triterpenes maslinic acid (MA), corosoic acid (CA), oleanolic acid (OA) and ursolic acid (UA) from.A method for the determination of MA, CA, OA and UA was established by HPLC. The contents of MA, CA, OA and UA were set as evaluation indexes, and the effects of ethanol volume fraction, extraction time and extraction times on the extraction efficiency were investigated by single factor and orthogonal experiment.MA, CA, OA, UA had good resolution of chromatographic peak, showing good linear relationships in the ranges of 0.568–3.408 μg (2=0.999 0), 0.582–3.492 μg (2=0.999 0), 0.488–2.928 μg (2=0.999 9), 0.720–4.320 μg (2=0.999 2), respectively. The recovery rates were 99.09% (RSD=1.57%), 99.76% (RSD=1.78%), 100.49% (RSD=1.66%) and 99.52% (RSD=1.75%), respectively. The optimal extraction technology conditions were: 95% ethanol extraction for 70 min, 3 times for extraction.The optimal extraction technology is reasonable, stable and feasible., and can be used to extract MA, CA, OA and UA from.
; orthogonal experiment; extraction technology; maslinic acid; corosoic acid; oleanolic acid; ursolic acid
R284.2
A
1005-5304(2021)06-0083-05
10.19879/j.cnki.1005-5304.202010251
云南省高等学校重点实验室建设计划(2019-2022年)
范源,E-mail:1647909799@qq.com
(2020-10-19)
(2020-11-30;编辑:陈静)
猜你喜欢
广东茶业(2019年2期)2019-06-18
中成药(2018年10期)2018-10-26
中成药(2018年8期)2018-08-29
中成药(2018年8期)2018-08-29
天然产物研究与开发(2016年1期)2016-06-05
医学研究杂志(2015年5期)2015-06-10
中国当代医药(2015年8期)2015-03-01
