
氧化石墨烯与剩余活性污泥聚合制备多孔碳材料及其电化学性能
2021-07-24 08:59肖弦徐文昊沈亮王远鹏卢英华
化工学报 2021年7期
肖弦,徐文昊,沈亮,2,王远鹏,2,卢英华,2
(1 厦门大学化学化工学院,福建厦门361005; 2 厦门市合成生物技术重点实验室,福建厦门361005)
引 言
剩余活性污泥作为一种城市污水处理厂的废弃物,传统的处理方法如填埋、燃烧或海洋倾倒,并未从根本解决问题。利用剩余活性污泥合成功能性材料是重要的资源化利用途径[1]。剩余活性污泥中含有大量的细菌生物质和丰富的有机成分,可考虑将其转化为碳材料。Feng 等[2]将城市污泥经700℃碳化得到三维蜂窝状分层结构的氮自掺杂碳材料,独特的3D 分层结构使得碳材料具有高比电容、优异的倍率性能及超长的循环稳定性,表明剩余活性污泥具有制备高性能电极材料的潜力。
石墨烯作为一种特殊的二维碳材料,具有比表面积大、强度高、导电性良好等优点,被认为是极具前景的超级电容器材料[3-5]。但由于强范德华力及剥离产生的高层间接触电阻,导致石墨烯不可避免及不可逆地发生重新堆叠或聚集,从而导致特定比表面积的大幅减少,并且最终导致特定电容较小(100~120 F/g)[6]。为了解决石墨烯的重新堆叠问题,将二维石墨烯自组装成三维结构,如水凝胶、气凝胶及水溶性泡沫,已被公认为是最有效的策略之一[7]。
研究表明氧化石墨烯(graphene oxide,GO)具有良好的生物相容性[8-9],且细菌个体优势使其能够阻挡GO 层间堆叠,在水溶液中二者聚集为三维石墨烯基材料[10-11]。与此同时,剩余活性污泥内含有还原能力的细菌(如地杆菌、希瓦氏菌)[12],可以将GO还原为导电的rGO,有助于电子的传递。此外,细菌也可以起到造孔剂的作用,增加材料的比表面积。剩余活性污泥中包含的大量富含氮细菌及有机物,还可作为杂原子掺杂剂的来源,从而提高材料的电化学性能。
因此,本文将采用剩余活性污泥(waste activated sludge,WAS)与GO 共聚法制备活性污泥石墨烯水凝胶(activated sludge graphene hydrogel,SGH)和干燥后的活性污泥石墨烯气凝胶(activated sludge graphene aerogel,SGA),进一步制备退火改性活性污泥法石墨烯气凝胶(annealing modified activated sludge graphene aerogel,ANSGA),并研究材料的电化学性能,探索其作为电极材料应用的可能性,为剩余活性污泥的资源化利用提供更加经济有效、环境友好的方式。
1 实验部分
1.1 主要试剂
实验中使 用 的300 目(通常,1 目×1 微米=15000)鳞片石墨分别购买于南京先丰纳米科技有限公司与国药集团化学试剂有限公司;高锰酸钾、30%(体积)过氧化氢溶液、无水乙醇、硝酸钠、氯化钠、盐酸、氢氧化钠、磷酸二氢钾、磷酸氢二钾、七水硫酸镁、碳酸氢钠、无水氯化钙、氯化钾、氯化铵、六水氯化镁、乙酸钠、硫酸钠购买于国药集团化学试剂有限公司;溴化钾购买于Thermo Fisher Scientific;酵母粉、蛋白胨购买于OXOID LTD;琼脂购买于鹭隆生物有限公司(分装);25%(质量)戊二醛购买于Acros Organics 公司。
1.2 实验
1.2.1 材料合成
(1)活性污泥石墨烯水凝胶(SGH)的制备。通过改进的Hummers 方法制得GO 悬浮液;剩余活性污泥来自福建省厦门市厦门前埔污处理厂二沉池,灰分含量约20%(质量),微生物组成以拟杆菌和变形杆菌为主[13],剩余活性污泥经7 d 驯化后[14],用于制备活性污泥石墨烯水凝胶(SGH)。
参考文献[15-16]方法制备SGH 所需乙酸钠培养基(SAM)。取NaH2PO41.3 g,Na2HPO4·H2O 14.5 g,用1 mol/L 的NaOH 及HCl 调节pH 至7.4,配制磷酸盐缓冲液(1 L)。取NaCl 9 g,用1 mol/L 的NaOH 及HCl调节pH至7.0,配制生理盐水(1 L)。取10 ml SAM放于厌氧管中,用氮气(N2)与二氧化碳(CO2)两种气体进行洗吹除氧。然后加入GO(0.2 mg/ml)和剩余活性污泥(MLSS 的浓度约为0.30 g/L),混匀,放入30℃的培养箱中进行静置培养,6 d 后,可以明显观察到黑色块状物与管壁完全分离,此时SGH形成。
(2)活性污泥石墨烯气凝胶(SGA)及退火改性活性污泥法石墨烯气凝胶(ANSGA)制备。SGH 浸泡于4 L装有超纯水的烧杯中进行透析,透析3 d,以除去残余在SGH 中的培养基,然后将其放于-20℃冰冻24 h,再于-40℃冷冻干燥1 d后,获得SGA。接着在氩气气氛中,将SGA 放入刚玉舟中,然后将其平放于水平管式炉中,以5℃/min 升温至700℃后,保持2 h后,在随炉冷却至室温后取出得到氩气退火改性的材料(ANSGA)。最后将ANSGA 浸泡于4 L 电解液中,该电解液为1 mol/L 硫酸溶液,浸泡时间为24 h,使得水和电解液可以得到充分的置换,再选取浸泡后的上述样品2 mg,放于前端剪去1 cm 的注射器开口端,垂直置于水平桌面,推动注射器活塞压制成片状,即可得到成型的材料样品。
自民国至今以来,虽以赞同“以诗为词”为主流,但仍存有个性化的反向论辩。施议曾在《词与音乐关系研究》中指出,苏轼“以诗为词”并没有很大的改革词风,其“独立抒情诗体只是其著作的一少部分,并没有彻底地打破‘诗庄词媚’、‘词为艳科’的传统观念”[12]230。崔海正在《东坡词研究中几个问题的再思考》中,则认为否定“以诗为词”这个命题比高举它好,因为“以诗为词”是从诗的立场来看词体,而不是立足于词本身来观察它的发展[4]330-331。
1.2.2 材料表征
(1)SGH 中rGO 化学性质测试。采用文献[17]方法洗除SGH 细菌并提取其中rGO,所得rGO 样品(命名为SGH-rGO)进行冷冻干燥,研磨为细腻的粉末状后将其压为表面平整的片状样品。利用如下仪器对其进行扫描分析:X 射线衍射仪(XRD,RigakuⅣ,日本Rigaku 公司),扫描范围为5°~60°,扫描速度10(°)/min,电压为40 kV,电流为30 mA;X 射线光电子能谱仪(XPS,Quantum 2000);傅里叶红外光谱仪(FTIR,Nicolate Avatar380,美国热电集团),扫描区域 为500~4000 cm-1;拉 曼 光 谱 测 试 仪(Raman,XploRA,法国HORIBA Scientific),采用50 倍物镜、532 nm 的激光激发测试。
(2)SGA 及ANSGA 的扫描电镜(SEM)分析表征。将获得的SGA 或ANSGA 样品黏附于导电胶上,并贴于样品台表面,干燥后将样品在喷铂仪下喷射30 s 后,立即上样观察,观察电压为15 KV。最后利用SEM附属配套的能谱分析仪对样品分析元素分析。
1.2.3 材料电化学性能测试 SGA 及ANSGA 材料的电化学性能测试为三电极体系。其中,1 mol/L 硫酸溶液作为电解液,饱和甘汞电极(SGE)作为参比电极,工作电极的面积质量负载量为2 mg/cm2,对SGA采取循环伏安法(CV)(扫描速率为10、25、50、75 及100 mV/s,扫描电压范围为-0.2~0.8 V)及恒流充放电(GCD)(电流密度分别为2、4、8、16、24、32、40 及48 A/g)的电化学测试手段。Pt 片上的SGA 或ANSGA作为工作电极,rGO 标样(抗坏血酸钠∶GO=3∶1(质量比),95℃,24 h)作为对电极进行电化学测试。材料的质量电容通过恒流充放电的放电时间由式(1)计算

式中,C(F/g)为比电容,I为横流充放电的放电电流,∆t为放电时间,m为工作电极中负载样品的质量,∆V为工作电极的电压范围。
2 结果与讨论
2.1 活性污泥石墨烯水凝胶形成过程及GO 化学性质变化的分析
图1 展示了SGH 形成的过程,可以观察到经过1 d 的时间,混合溶液出现分层现象,并且从最开始的棕褐色转变为黑色,表明GO 可能被污泥中的细菌还原而除去表面大部分的含氧官能团,从而产生了疏水的rGO[10,18]。
经过3 d 的培养后,可以发现黑色产物纵向边缘沉降到厌氧管的1/3 处,且横向边缘也开始收缩,此时由于GO 的含氧官能团被去除,片层间的静电排斥作用力减小,片层相互靠近,发生π-π 堆叠现象[19],因此黑色产物继续沉降,可观察到SGH的基本雏形。直至到第6天,黑色产物完全脱离试管壁,且边界清晰分明,表明最终形成了三维活性污泥石墨烯水凝胶,而凝胶周围的残留溶液几乎是无色透明的,表明GO和活性污泥都已进入凝胶固体中。
图2(a)、(b)为GO 和SGH 中rGO 的C 1s 分 峰的XPS 光谱图,可以观察到GO 的XPS 光谱分别在284.5、286.6、287.2 和288.9 eV 处显示出C—C 骨架、C—O 键、C O 键和O—C O 键的特征峰。与原始GO 相比,还原后GO 的XPS 谱图发生了较大的变化。C—O 的相对含量从20.7%下降到15.7%,C O相对含量从16.8%下降到0,O—C O 的相对含量从14.1%下降到11.3%,而C—C 的相对含量则从48.4%上升到73.0%。该结果证实是细菌还原引起了GO部分含氧官能团的去除。
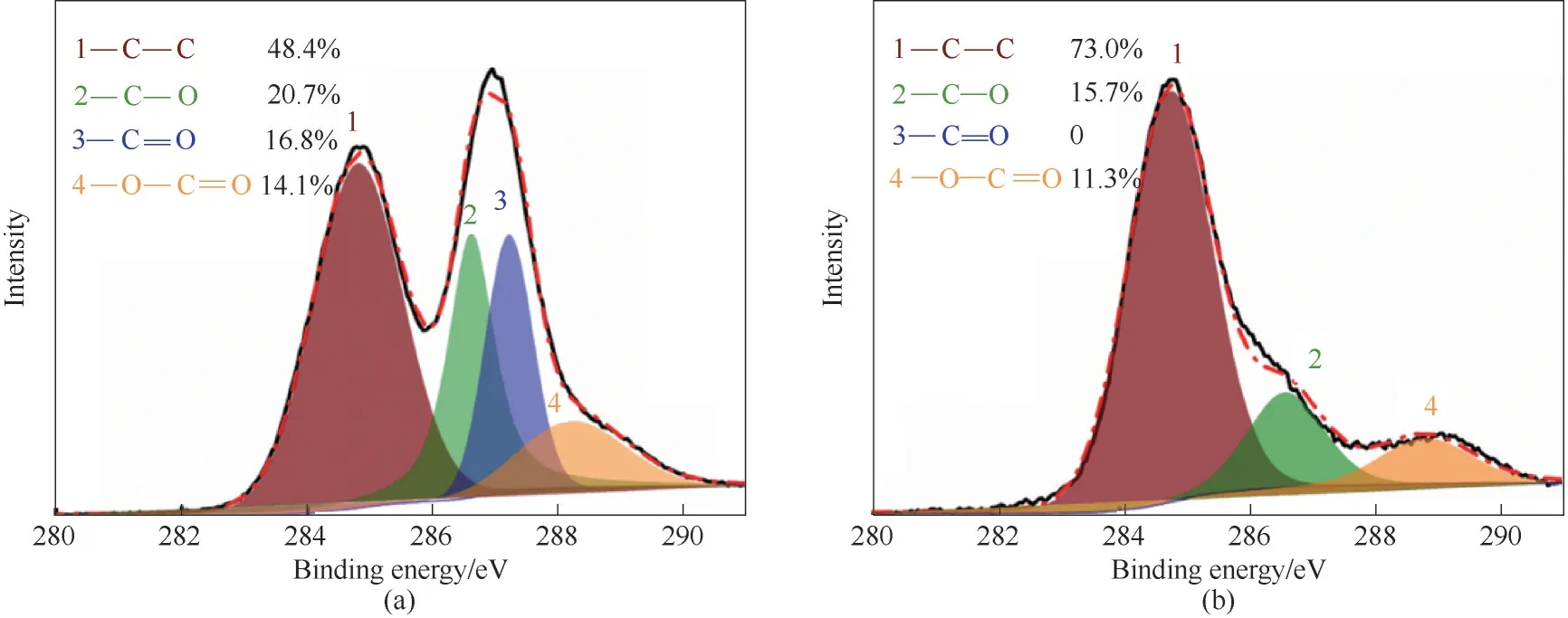
图2 GO(a)和SGH中rGO(SGH-rGO)(b)的XPS光谱图Fig.2 XPS C 1s spectra of GO(a)and SGH-rGO(b)
Raman谱图可用于表征石墨烯及石墨烯相关材料的晶体结构、无序和缺陷。从图3(c)看出两种样品均表现出两个强峰:约1560 cm-1(G波段)的单个尖锐信号和约1350 cm-1(D 波段)的附加信号。D 波段与G 波段的强度比(ID/IG)可用于反映石墨烯中缺陷之间的平均距离(LD),增加的ID/IG值可作为指示GO被成功还原的信号。经过D峰及G峰的峰高比值计算 可 知,GO 与SGH 中rGO 的ID/ IG分 别 为0.95 和1.03。结果表明还原后的GO表面缺陷减少,这主要是归因于细菌还原后,GO 表面的含氧官能团大量减少,修复了一些存在于石墨烯片层表面的非永久性缺陷,同时也保留了永久缺陷[22],改变了原本石墨烯完美的结构,间接证明GO被细菌还原了。
综上所述,通过XPS 定量考察GO 还原前后的含氧官能团种类分布、FTIR 定性考察GO 还原前后C、N、O、S 等官能团的变化、XRD 考察层间距、Raman 考察还原前后晶格缺陷程度,最终证明了弱导电GO 被还原为强导电的rGO 及剩余活性污泥中微生物的还原作用,从而具备了应用于电化学领域的潜力。
2.2 活性污泥石墨烯气凝胶的亲水性和O、N 自掺杂结构
图4(a)、(b)显示了水在SGA 上散布,而水接触角在9 s 内接近0°,表明SGA 具有亲水性。材料的亲水性促进了水性电解质的吸附。
图4(c)~(e)中的FTIR 谱图和XPS 谱图结果一致,证明了细菌还原后SGA-rGO 中存在亲水性官能团(—COOH 和C—O)。此外,图5 中的XPS 谱图证明了SGA 中存在C、O 和N 元素。元素分析结果也证实了SGA 中存在N,表明各组SGA 样品中N 的百分比在3%~4%(atom)之间。含氧基团的存在和少量N官能团的存在为制备相应的超级电容器提供了必不可少的亲水性和赝电容[23],故SGA 具有制备活性电极材料的可能性。但SGA 掺氮来源于细菌自身的蛋白质等含氮物质,细菌生物质的灰分含量约为20%,在细菌与GO 聚合以及SGH 冻干过程中这部分灰分并没有得到有效去除。考虑到灰分对于电极材料性能的重要影响,SGA 还需经过物理或化学处理后才能用作电极材料。

图3 GO与SGH-rGO的红外光谱图(a),XRD谱图(b),拉曼光谱图(c)Fig.3 FTIR spectra(a),XRD patterns(b),and Raman spectra(c)of GO and SGH-rGO
2.3 活性污泥石墨烯气凝胶及退火改性活性污泥法石墨烯气凝胶的结构及形貌对比
孔隙结构是影响材料性能的重要因素。通过分析两种材料在低倍镜下的扫描电镜图,对比退火后材料在形貌结构上发生的改变。
如图6(a)所示,SGA 内部疏松多孔,出现许多孔径为10µm 左右的明显大孔,是一种典型的大孔材料。除此以外,材料内部也有少量大小与细菌体积相似的2µm左右的孔径,说明细菌可以在凝胶形成过程中起到造孔剂的作用,形成了以微米级大孔为主的多孔结构。然而,对电极材料电容性能影响更大的是微孔(<2 nm)和介孔(2~50 nm),SGA由于孔径过大,电解质不能有效扩散到材料内部发生吸附作用,实际上不利于储能,如SGA 用作电极材料则需进一步加工形成微孔和介孔[24]。在同样放大倍数下对比退火后的材料ANSGA[图6(d)]可以发现,ANSGA 仍以微米级孔洞为主,但内部结构比起SGA更加致密且形成了更多不规则的蜂窝状孔洞,更多孔径的形成能为电解质的传输提供更丰富的通道,这说明在氩气中高温退火的操作对于提升材料的孔隙率起到了关键的作用。主要原因是当温度处于100~200℃的时候,整个SGA 材料内部会伴随水分子的蒸发发生一定的聚合;随着温度逐渐升高至200~400℃,存在于细菌及胞外聚合物(extracellular polymeric substance,EPS)中的氨基酸会受热分解产生NH3,并逐渐扩散穿过碳层,从而产生孔隙达到造孔的目的[25]。温度继续升高至400~700℃,SGA 中的含氧官能团受热分解,与碳骨架刻蚀反应生成CO、CO2以及水蒸气等小分子气体[26]。此外,当温度超过600℃时,N 原子会转化为吡啶N 和吡咯N 等具有较高热稳定性的官能团,并且部分N 原子可能进入碳的内部结构形成石墨N,从而形成氮掺杂的材料[26]。通过测出的EDS 结果表明,ANSGA 的掺N 含量可以达到5.61%(质量)。最后,NH3、CO2、水蒸气在700℃保持2 h 的过程中进一步进行物理活化造孔,从而实现一体化的造孔与掺氮。
2.4 ANSGA的电化学性能

图4 0 s时SGA的接触角(a);9 s时SGA的接触角(b);GO和SGA-rGO的FTIR光谱图(c);GO(d)和SGA-rGO(e)的XPS C 1s光谱图Fig.4 Contact angle of SGA at 0 s(a);Contact angle of SGA at 9 s(b);FTIR spectra(c)and XPS C 1s spectra[(d),(e)]of GO and SGA-rGO

图5 SGA的XPS谱图Fig.5 XPS survey spectra of SGA
通过循环伏安测试、恒流充放电测试及循环稳定性测试来测定ANSGA 的电化学性能,并根据GCD 曲线进一步计算出样品在不同电流密度下的比电容量。图7(a)为ANSGA 在不同的扫描速率下的循环伏安(CV)图,可以明显的看到ANSGA 的CV曲线分别位于0.2~0.4 V 和0.4~0.6 V 出现了一对明显的氧化还原峰,这属于典型的赝电容特征,这是由于掺杂N原子发生了法拉第氧化还原反应为电极材料提供了部分的赝电容。除此以外,CV曲线展现出显著的对称性,这说明电极的氧化还原反应具有非常好的可逆性,充放电完全。随着扫描速率的增加,CV 曲线的面积增大,但形状仅发生了微弱的变形,这表示ANSGA具有非常优异的倍率性能和离子传输特性,这对于材料的电化学应用是十分有利的。
材料优异的电化学性能在GCD 曲线中也得到了相应的印证。在图7(b)中可以看到,不同电流密度下的GCD 曲线在0.2~0.4 V 的范围内充放电曲线都出现了明显的平台,这与双电层电容所呈现的对称等腰三角形极为不同,说明发生了氧化还原反应。且GCD 曲线中放电曲线与其对应的充电曲线几乎对称,说明充电时间与放电时间近似相等,显示出材料优异的电化学可逆性[27-28]和良好的库仑效率,今后可以通过长循环图对库仑效率进一步定量分析[29-30]。

图6 SGA[(a)~(c)]和ANSGA[(d)~(f)]的扫描电镜图Fig.6 SEM images of SGA[(a)—(c)]and ANSGA[(d)—(f)]
根据GCD 曲线并通过公式计算出ANSGA 在不同电流密度下的比电容值,如图7(c)所示,在2 A/g的电流密度下初始比电容值为174 F/g,相较于先前报道的外源掺氮石墨烯基碳材料的比电容,该值接近甚至更高,其掺氮量(5.61%)也高于部分外源掺氮的石墨烯碳材料[31-36]。而本研究中自渗氮的方式比起外源渗氮(如DMF),在操作上更加简单、成本更加低廉且不易产生环境污染。在4、16、24 及48 A/g 的电流密度下,比电容分别为165、147、142及130 F/g,其电容保持率可达到初始的74.7%(48 A/g),高于其他引入金属离子及同样含有细菌的掺氮石墨烯基碳材料[34-37](表1)。此外,该电极在21 A/g 的电流密度下经过12000 次循环充放电测试后,比电容值仍能保持初始的72%[图7(d)],展现了其优异的循环稳定性。
材料优异的电化学性能与合适的孔径大小及分布有密切关系,当电流密度增大时,拥有大孔径的电极仍快速地传输电解质,最终保证材料的电化学性能维持平稳。此外,N 原子的掺杂使得石墨烯表面上产生缺陷,石墨烯基面上的缺陷孔洞可以作为一种快速输送离子或电解质路径,从而提升材料电化学性能。基于电容量的提升、优异的倍率性能、离子传输特性和循环稳定性,ANSGA 作为超级电容器的电极材料具有较大的潜力。
2.5 高温退火对SGA电化学性能的影响评估
对退火前后的SGA 电化学性能进行比较,以探究退火操作对于材料电化学性能的影响。
CV 曲线的面积大小在一定程度上体现了材料比电容的大小,如图8(a)所示分别为SGA 与ANSGA两种样品在50 mV/s 条件下的CV 曲线,从中可以明显观察到ANSGA的CV曲线包围的面积远大于SGA的面积。因为两种样品的面积相差较大,为了更清晰地观察到SGA 的CV 曲线并且精准地对比两者的区别,图8(b)展示了SGA 在不同扫描速率下的CV 曲线,从图中可以观察到SGA 虽然同样具有一对氧化还原峰,但是随着扫描速率的增加,曲线的形状发生了显著的变化,并且对称性变得不如ANSGA 完美,说明SGA的倍率性能远差于ANSGA。

图7 基于退火后的SGH的电化学性能:循环伏安测试(a);不同电流密度下恒流充放电测试(b);不同电流密度下的比电容(c);21 A/g下的循环稳定性测试(d)Fig.7 Electrochemical performance of ANSGA:CV curves(a),GCD curves(b),specific capacitance at different current densities(c),and cyclic stability test at a current density of 21 A/g(d)
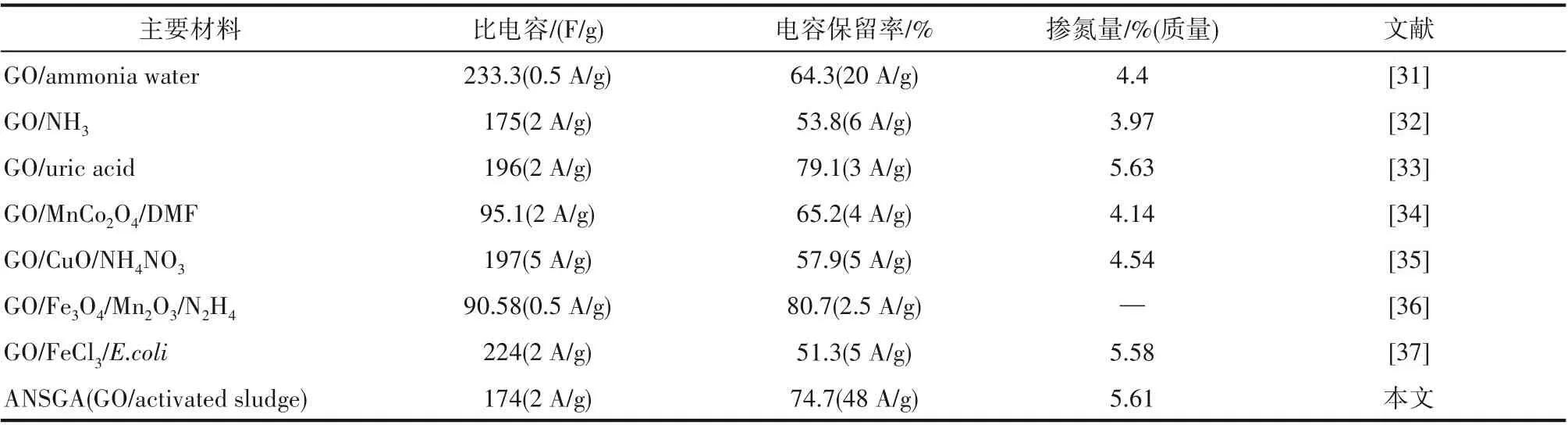
表1 石墨烯基碳材料的比电容、电容保留率及掺氮量的对比Table 1 Comparison of specific capapcitance,capacitance retention and N-doped content of graphene-based carbon material
此结果在GCD 曲线对比中也得到了验证,如图8(c)所示为2 A/g 的电流密度下两种材料的GCD 曲线,可以观察到ANSGA的氧化还原峰的电位平台明显较宽,且充放电时间长达102 s,显著地高于SGA。并且ANSGA 的GCD 曲线相较于SGA 的更加对称,意味着SGA 材料的内阻较大,导致其库仑效率不高。
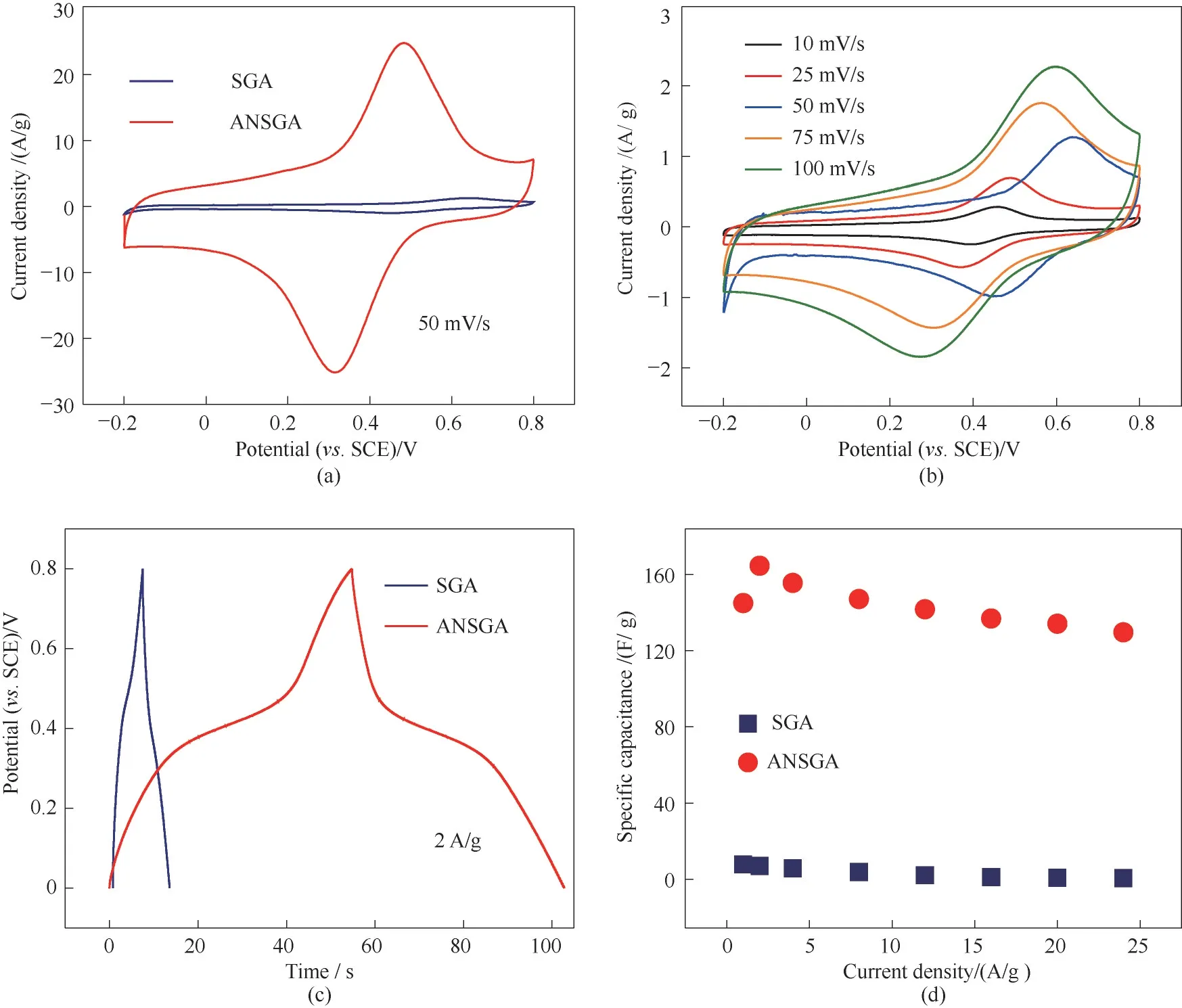
图8 基于SGA及ANSGA的电化学性能:50 mV/s下循环伏安曲线(a);SGA的循环伏安测试(b);2 A/g下恒流充放电曲线(c);不同电流密度下的比电容(d)Fig.8 Electrochemical performance of SGA and ANSGA:CV curves at a scan rate of 50 mV/s(a),CV curves of SGA(b),GCD curves at a current density of 2 A/g(c),and specific capacitance at different current densities(d)
经过计算可得在电流密度为2 A/g 的条件下,ANSGA 的比电容值(174 F/g)比SGA(8 F/g)约高22 倍[图8(d)],这说明氩气中退火的操作极大地提高了材料的比电容。原因主要是ANSGA 的孔隙率远高于SGA,因此更有利于在高电流密度下快速传输电解质,并储存部分电解液继续发生作用。此外,在氩气环境下进行退火操作时,活性污泥中存在多种细菌以及EPS 如多糖、蛋白质和腐殖质等物质,在700℃高温且无O2环境下退火后,活性污泥中的含碳物质会发生脱水碳化,以无定形碳的形式存在于材料表面上,而碳本身就是具有优异导电性的材料,可以有效减小内阻,帮助快速传递电子[38]。另一方面,活性污泥中的大量复杂有机物在高温下可分解为小分子气体,从SGA 中逸出,既实现了造孔又增加了材料的孔径从而扩大了比表面积,为电子材料提供了更多的活性中心。
综上所述,高温退火操作有助于提高材料的碳含量及孔隙率,材料内细菌、EPS 及含氧官能团在高温下分解为气体,从而实现掺氮与造孔,进而增强材料的电化学性能。因此,氩气中高温退火的操作对于提升材料的电化学性能具有关键作用。
3 结 论
(1)剩余活性污泥与氧化石墨烯自发共聚制得生物石墨烯水凝胶(SGH),干燥后得到活性污泥石墨烯气凝胶(SGA),能够同时实现多孔碳材料的绿色制备以及剩余活性污泥的资源化利用。
(2)剩余活性污泥中的微生物将弱导电性的GO还原为强导电的rGO。
(3)退火改性活性污泥法石墨烯气凝胶(ANSGA)具有优异的电化学性能,包括优异的倍率性能、离子传输性能和循环稳定性,其电容保持率可达到初始的74.7%(48 A/g),即使在21 A/g 的电流密度下循环12000 次后电容保持率也超过72%,高于先前报道的其他石墨烯基碳材料。
(4)在氩气(700℃,2 h)中退火后的ANSGA 在2 A/g 的电流密度下比电容值较退火前提高22 倍,达到174 F/g,表明高温退火对提高ANSGA 电化学性能有关键作用,如ANSGA进一步加工微孔结构及去除灰分含量,可能发挥电极材料的潜在作用。
猜你喜欢
区域治理(2022年3期)2022-03-05
蓄电池(2022年1期)2022-02-25
皮革制作与环保科技(2021年8期)2021-11-28
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
化工管理(2021年7期)2021-05-13
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
当代化工(2019年8期)2019-12-13
表面工程与再制造(2019年6期)2019-08-24
绿色科技(2019年12期)2019-07-15
科技创新与应用(2017年11期)2017-04-27
