
面肩肱型肌营养不良1型1例
2021-07-24 03:42罗雅尹梁战华周丽娜宋春莉
大连医科大学学报 2021年3期
罗雅尹,王 哲,梁战华,周丽娜,周 密,宋春莉
(大连医科大学附属第一医院 神经内科,辽宁 大连116011)
1 临床资料
患者,男,29岁。因“被发现肩胛凸起10年,双上肢抬举无力6年余”于2018年11月2日入大连医科大学附属第一医院神经内科诊治。10年前无意中被发现肩胛凸起,未在意。6年前发现双上肢上举无力,以右侧为重,表现为不能抬起重物,左侧无力症状不明显,不影响日常生活。3年前因上肢力弱而锻炼时发现左上肢不能举重,抬举较前费力(图1)。上述症状持续存在并缓慢加重,现无法自行完成脱上衣、洗头、梳头等动作,屈肘、抓持等力量可;病程中未出现下肢无力、肢体麻木,无视物模糊及尿便障碍等,未行特殊诊治,体重无明显下降。

图1 患者双上肢抬举无力,右侧更严重Fig.1 Patient's upper limbs were weakin lifting, and the right side was more severe
既往史及个人史:无特殊。家族史:母亲幼年右耳失聪,右眼外斜视,在38岁时诊断糖尿病。
体格检查:神清语明,精神智能正常。面部表情少,双眼闭合稍无力,左侧为重,左侧鼓腮轻微漏气,转颈耸肩肌力正常,余颅神经大致正常。翼状肩胛,双侧冈上、冈下肌、前锯肌、胸大肌不同程度萎缩(见图2、图3),余肌肉未见明显萎缩。双上肢远端肌力正常,近端肌力4级,双下肢肌力正常,四肢肌张力正常,双侧肱二、三头肌腱反射、桡骨膜反射对称引出,双侧膝、跟腱反射对称引出。双侧病理征(-),跖反射中立位。双足爪型趾改变。

图2 翼状肩胛、肩胛带肌萎缩Fig.2 Atrophy of scapular girdle and pterygoid scapula

图3 肌肉萎缩(箭头所指)Fig.3 Muscle atrophy (arrows)
辅助检查:血清肌酸激酶168 IU/L(参考值55~170 IU/L),余血生化、风湿免疫全套、甲功、肿瘤标志物、肝炎梅毒HIV检查正常。头核磁、颈椎核磁、心电图、腹部及泌尿系彩超、胸部CT均正常。
鉴于患者青少年期起病,缓慢进展,慢性病程,主要表现为肩胛带肌萎缩无力,面部表情少,无感觉障碍,首先考虑为肌肉病,面肩肱型肌营养不良可能性大,故行肌电图检查协助诊断。肌电图结果如下:(1)感觉神经传导速度正常(双侧正中神经、尺神经、桡神经、前臂内外侧皮神经、胫神经、腓浅神经、腓肠神经);(2)运动神经传导速度:双侧胸长神经、副神经波幅下降;双侧正中神经、尺神经、桡神经、腋神经、肌皮神经、胫神经、腓总神经均正常;(3)F-波:正中神经、胫神经均正常;(4)针电极肌电图(上下肢、斜方肌、前锯肌、胸锁乳突肌、胸段脊旁肌):所检肌肉安静状态下可见1~2处自发电位,右前锯肌可见CRD;小力收缩时可见宽大动作电位和窄小动作电位混合存在,部分肌肉平均时限增宽>20%;大力收缩呈单纯相或混合相,部分肌肉可见早募集现象(图4~5)。提示肌源性损害和神经源性损害混合存在。这给电生理和临床医生带来了诊断上的困惑,为进一步明确诊断,2018年11月13日委托基因检测公司行基因分析。基因分析结果:(1)受检者(先证者):检测到一条4号染色体(4q35)亚端粒区多态性EcoR Ⅰ/p13E-11片段长度缩短至25.5 kb(正常参考值38~300 kb),为4qA型;另一条4q35亚端粒区多态性EcoR Ⅰ/p13E-11片段>38 kb,为4qB型;同时,存在10q-4q的易位。符合面肩肱型肌营养不良(FSHD)的诊断。(2)受检者(父亲):检测到的两条4号染色体(4q35)亚端粒区多态性EcoR Ⅰ/p13E-11片段长度均>38 kb(正常参考值38~300 kb),为4qB/4qB型;同时存在10q-4q的易位。(3)受检者(母亲):检测到一条4号染色体(4q35)亚端粒区多态性EcoR Ⅰ/p13E-11片段长度缩短至25.5 kb(正常参考值38~300 kb),为4qA型;另一条4q35亚端粒区多态性EcoR Ⅰ/p13E-11片段>38 kb,为4qB型;符合FSHD的基因结构变异。该病例最终经基因检测确诊为面肩肱型肌营养不良1型。患者于2018年11月20日出院,未予特殊药物治疗,嘱其补充营养,适当活动锻炼。2年后电话随访,双上肢抬举无力未见好转,较前无明显加重。
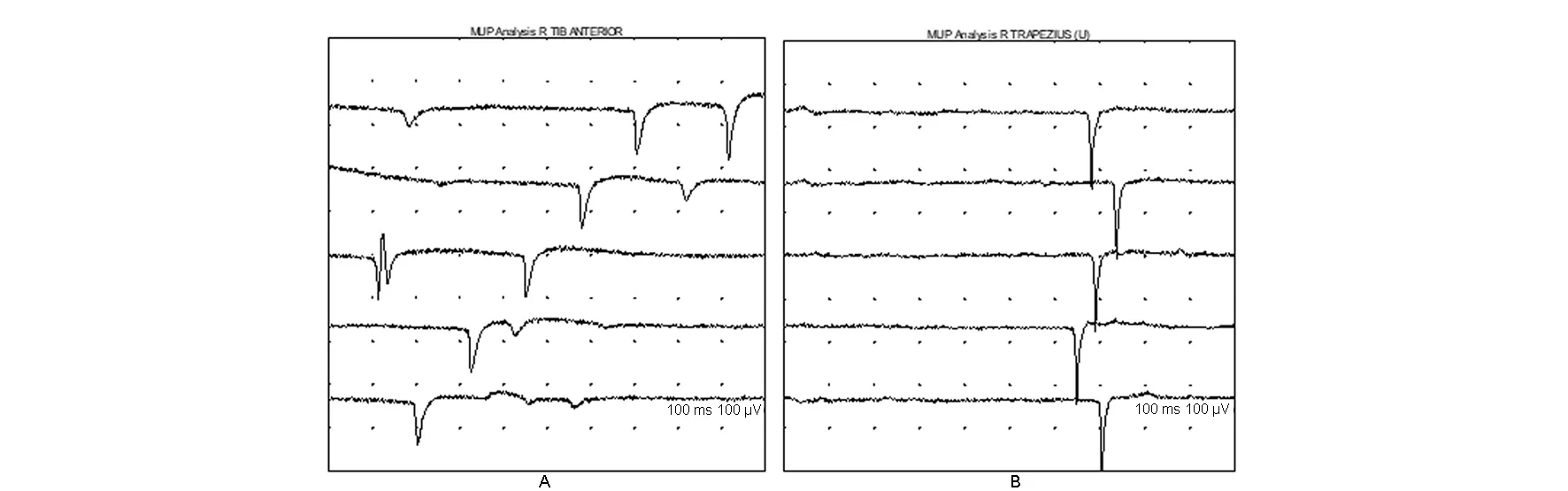
图4 右侧胫骨前肌(A)、右侧斜方肌(B)安静状态下可见正锐波Fig.4 Right anterior tibialis (A) and right trapezius (B) showed positive sharp waves at rest in EMG
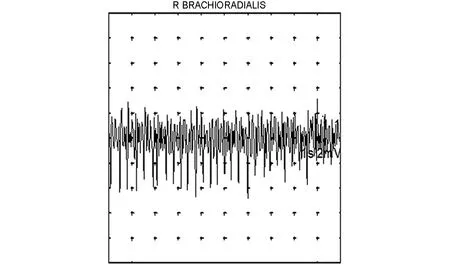
图5 右侧肱桡肌大力收缩可见早募集现象Fig.5 Right brachioradialis contraction showed early recruitment phenomenon in EMG
2 讨 论
本例患者临床表现符合面肩肱型肌营养不良(FSHD),作为一种遗传性肌肉疾病,肌电图通常表现如下:感觉神经传导速度完全正常;在疾病早期,运动神经传导速度也正常,但随着疾病进展,伴随肌肉萎缩可出现CMAP波幅下降,但传导速度正常[1]。本例患者病程10余年,肩胛带肌萎缩,双侧胸长神经和副神经CMAP波幅下降。针电极肌电图通常示肌源性损害,安静状态下可有纤颤电位、正锐波等自发电位的出现,且早期相对较多,随着病程延长,自发电位数量相对减少[2];轻收缩表现为MUP时限缩短、波幅减低且多相波增多,有早募集现象,重收缩呈病理干扰相[3]。本病例所检肌肉可见1~2处自发电位,符合慢性遗传性肌肉病的特点。但是本病例肌电图的疑点难点在于轻收缩时出现了MUP时限增宽、波幅增高,部分肌肉平均时限增宽>20%,提示神经源性损害,且重收缩夹杂着高波幅MUP,很容易误认为单纯相,需要有经验的电生理医生加以识别,注重重收缩整个过程募集情况,辨认早募集现象。
有研究指出多数炎性肌病肌电图可合并出现神经源性损害,认为是同一种疾病的表现,如炎性肌病中肌炎和血管炎性周围神经病变等[4]。也有研究报道伴有神经源性损害的非炎性肌病,如脂质沉积性肌病、中央轴空病,目前认为是由长期慢性肌病导致失神经支配所致[5]。FSHD进展缓慢,既往曾有少数案例发现面肩肱型和肢带型肌营养不良出现肌源性损害合并神经源性损害[6]。本例患者病程10余年,符合肌肉疾病经过长时间慢性失神经损害,导致肌电图表现变得复杂,在疾病过程中如果出现轴索末梢损害后神经再支配,可表现为宽大的 MUP,或宽大与缩窄的MUP混合存在,需要注意。
FSHD临床分型包括FSHD1型和FSHD2型,前者多见,与4q35区域D4Z4串联重复序列缺失有关,4q35区域DNA低甲基化启动表观遗传效应,使D4Z4串联重复序列内DUX4基因去抑制致异常表达,导致多种肌细胞损害效应。后者与DNA甲基化调控基因-SMCHD1基因突变有关[6-7]。本例患者基因型符合FSHD1型。
肌电图有助于神经肌肉疾病的诊断和鉴别诊断,是鉴别神经源性损害和肌源性损害最方便且有价值的检测手段,不但能够鉴别损害类型,而且能够提示损害范围。FSHD肌电图通常表现为肌源性损害,但是本病例揭示了FSHD肌电图可出现神经源性损害,提示我们病程较长的肌肉病可能继发神经损害,肌电图表现肌源性损害和神经源性损害共存,在电生理医生规范操作的基础上,需要结合临床表现综合判断,基因检测可确诊。
目前该病缺乏有效治疗方法,以对症支持治疗为主,物理疗法和康复治疗对维持活动功能很重要,建议增加营养、适当活动,FSHD患者的预后相对较好,部分患者寿命可接近正常生命年限。
猜你喜欢
康复(2022年25期)2022-11-18
基层中医药(2022年7期)2022-11-17
中国运动医学杂志(2022年8期)2022-10-14
临床骨科杂志(2022年4期)2022-08-30
现代仪器与医疗(2022年2期)2022-08-11
骨科临床与研究杂志(2021年4期)2021-07-14
国际骨科学杂志(2021年2期)2021-04-21
上海医药(2017年2期)2017-03-01
中国实用医药(2016年26期)2016-11-07
中国实用医药(2016年1期)2016-01-11
