
不同液相色谱-串联质谱法检测标准测量不确定度的评定
2021-07-22 00:43韩帅兵张耀中李向阳吴亚玉
农药科学与管理 2021年6期
韩帅兵,张耀中,于 淼,薛 雯,李向阳,吴亚玉,周 力
(山东省农药检定所, 山东 济南 250100)
随着广大人民群众及政府部门对农产品质量安全重视程度的不断加强,各检测机构承接的农产品中尤其是蔬菜水果中农药残留检测任务不断增加,如何提供更加科学、准确的检测数据也成为各实验室关注的焦点。测量不确定度代表着检测值的可信区间,是衡量实验室检测质量的重要指标,尤其是检测结果在限量临界值区间时,不确定度更是合格性判定的重要参考依据[1-3]。因此,不确定度的评定逐渐受到各个实验室的重视。随着不确定度评定相关标准及指南的更新或发布[4-5],不确定度的评定更加有据可循,测量不确定度的评定工作也相应成为各实验室检测工作的重要一环。当前,液相色谱-串联质谱法作为一种应用越来越普遍的农药残留检测方法,关于它的测量不确定度评定的研究虽有所报道[6-8],但这些报道大多不是建立在标准规定方法之上,对检测实验室具体工作参考意义有限。
2021年3月,新的国家标准GB 23200.121-2021《食品安全国家标准 植物源性食品中331种农药及其代谢物残留量的测定 液相色谱-质谱联用法》发布[9],此方法是建立在QuEChERS方法基础上的多残留检测方法,与原国家标准GB/T 20769-2008《水果和蔬菜中450种农药及相关化学品残留量的测定 液相色谱-串联质谱法》[10]相比,该方法用料量少、操作简便,且涉及了很多原标准未涉及的农药代谢物,一定程度上来说,可以替代原国标。这两种检测方法在前处理步骤上存在明显差异,那么,在测量不确定度的评定过程及结果上是否也存在明显差异,目前还没有相关文献报道。
本研究以检测大白菜中吡虫啉残留量为例,按照标准规定的具体操作步骤及计算方法进行检测,依据不确定度评定相关标准及指南[4-5],分别评定了在相同检测条件下,两种检测方法测量结果的不确定度,比较了二者在不确定度组成、最终结果、贡献度分布及关键因素上的区别,并分析了产生原因,为标准方法的比较提供了数据依据,同时也为农药残留检测实验室提供了不确定评定的参考范例。
1 实验部分
1.1 主要仪器与材料 超高效液相色谱-串联质谱仪(ESI);氮吹仪(N-EVAP112);旋转蒸发仪(RV10 Control);移液器;石墨炭黑氨基SPE小柱(Carbon/NH2SPE,500mg,6mL);0.22μm有机滤膜、量筒、容量瓶等。
1.2 主要试剂 乙腈,色谱纯;甲醇,色谱纯;甲苯,分析纯;氯化钠,分析纯;无水硫酸镁,分析纯;柠檬酸钠,分析纯;柠檬酸氢二钠,分析纯。
1.3 样品前处理 样品分别按照两种标准规定的方法进行前处理。
1.3.1 GB/T 20769-2008前处理 提取:准确称取20g(精确至0.01g)样品于80mL离心管中,加入40mL乙腈,15 000r/min下匀浆1min,加入5g NaCl再匀浆1mL,离心后取上清液10mL,40℃氮吹至1mL后待净化。
净化:用5mL的V(乙腈) : V(甲苯)= 3:1混合液预淋Carbon/NH2柱后,加入待净化液,用2mL上述混合液分3次洗涤离心管,将洗涤液转入柱中,再用25mL淋洗液洗脱,收集所有洗脱液,旋转浓缩近干,用5mL乙腈定容,过0.22μm有机滤膜后待测。
1.3.2 GB 23200.121-2021前处理 提取:准确称取10g(精确至0.01g)样品于50mL离心管中,加入10mL乙腈、4g无水硫酸镁、1g氯化钠、1g柠檬酸钠、0.5g柠檬酸氢二钠及1颗陶瓷均质子,盖上离心管盖,剧烈震荡1min 后4 200r/min离心5min。
净化:吸取6mL上清液至内含900mg无水硫酸镁、150mgPSA的离心管中,涡旋混匀1min。4 200r/min离心5min,吸取上清液过0.22μm有机滤膜后待测。
1.4 样品检测 前处理后的样品经UPLC-MS/MS检测,两种检测标准采取相同的仪器方法,参考仪器条件如下:
色谱柱:Shim-pack XR-ODS Ⅲ(2.0mm I.D.×50mm,1.6μm)
流动相:A:甲醇,B:5 mM醋酸铵+0.02%甲酸水溶液;柱温:40℃;进样量:0.5μL
流速及梯度:(表1)
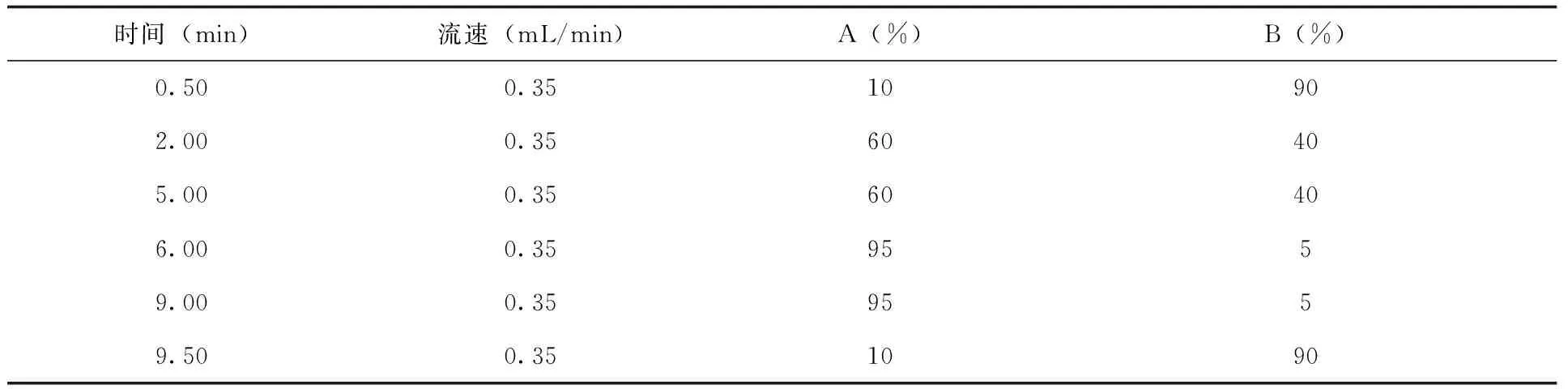
表1 流动相流速及梯度设置
质谱仪条件:离子源:ESI源,正离子模式;检测方式:MRM;电离电压:4.5kV;碰撞气及压力:Ar,0.23MPa;雾化气流速:3.0L/min;检测器电压:1.74kV;干燥气流速:10.0mL/min;加热气流速:10.0mL/min;定量离子:256.10-209.10,碰撞电压-14V;定性离子:256.10-175.10,碰撞电压-16V。
1.5 数学模型的建立 根据两种检测标准规定的方法,两种方法的残留量(mg/kg)计算分别见公式(1、2)。
(1)
(2)
其中,公式(1)为GB/T 20769-2008计算公式,公式(2)为GB 23200.121-2021计算公式。式中,ω为样品中被测农药组分的含量(mg/kg);Ai为样品中被测农药组分的峰面积;As为标准溶液中被测农药组分的峰面积;V1为提取溶液总体积(mL);V2为吸取出用于检测的提取溶液体积(mL);V3为样品定容体积(mL);m为样品取样量(g);ρ为标准溶液中农药的含量(mg/L)。
2 结果与讨论
2.1 不确定度来源分析 通过对检测过程的分析,两个标准的检测都有4个主要步骤:标准溶液配制、称样、前处理和上机检测,据此可将引入不确定度的来源细化为:所购买标准溶液、标准溶液配制、称样、前处理过程、仪器稳定性。另外,在实际检测工作中,还需要考虑方法精密度对测定结果的系统性影响,由此可以得到不确定度来源分析鱼骨图(图1)。

图1 不确定度来源分析鱼骨图
其中,对于GB 23200.121-2021来说,样品前处理过程中并不包含分取和定容的过程。综合公式(1、2)及(图1),可以判定检测结果的不确定度主要来源包括:标准溶液质量浓度(ρ),样品进样峰面积(Ai),标准溶液峰面积(As),样品的最终定容体积(V),样品称样量(m)及方法精密度(frec),由此可根据公式(3)得到合成不确定度urel(ω):
即:urei(ω)=
(3)
2.2 相对不确定度的评定
2.2.1 标准溶液引入的不确定度 两种检测方法所用标准溶液均为用购买的吡虫啉有证标准溶液稀释后得到,因此,由标准溶液引入的不确定度是相同的,包括所购标准溶液urel(ρ1)和配制过程引入urel(ρ2),均为B类评定。
2.2.1.1 购买标准溶液引入的不确定度 本研究所购买吡虫啉标准溶液质量浓度为1 000mg/L,标准物质证书显示其扩展不确定度为7mg/L(包含因子k=2),则所购标准溶液引入的相对不确定度为:urel(ρ1)=0.35%。
2.2.1.2 标准溶液配制引入的不确定度 本研究最终所用质量浓度为0.2mg/L的标准工作溶液是使用质量浓度为1 000mg/L的标准溶液用乙腈2次稀释后配制而得:先用1mL A级分度吸量管移取0.25mL 1 000mg/L标准溶液,用乙腈定容到25mL,配制得1.0mg/L标准溶液;再用1mL A级分度吸量管移取1.0mL 1.0mg/L标准溶液,用空白样品溶液定容到5mL,得到0.2mg/L的标准工作溶液。在标准溶液的配制过程中,引入不确定度的因素应该包括温度变化对试剂膨胀的影响,及配制过程中所使用玻璃仪器的允差及读数误差。
实验室温度一般控制在(20±5)℃,乙腈的膨胀系数为0.001 37℃-1,服从矩形分布,由此,由温度效应引入的相对不确定度可由公式(4)计算而得:
(4)
配制过程中所用玻璃仪器引入的不确定度主要由允差和读数重复性偏差组成,每个分量可用公式(5)计算:
(5)
式中aL为玻璃仪器的最大允许误差或读数重复性偏差,其中,1mL A级分度吸量管精度较高,读数重复性引入不确定度可以忽略不计,允许误差为±0.008mL[11];25mL容量瓶允许误差为±0.03mL,重复性偏差为±0.01mL;5mL容量瓶允许误差为±0.02mL,重复性偏差为±0.01mL;服从三角形分布,k取 ;V为所用体积。由此,在标准溶液配置过程中每次使用玻璃仪器由误差、偏差及温度引入的不确定度(表2)。
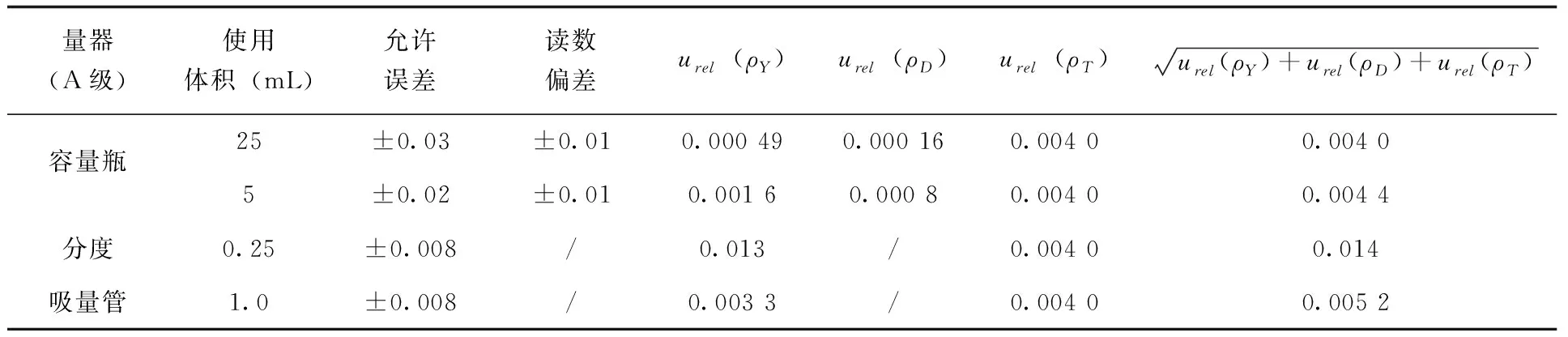
表2 标准溶液配制中玻璃仪器使用引入的不确定度
将上述不确定度合并,得到标准溶液配制过程引入的不确定度urel(ρ2)公式(6)。
(6)
据此,计算标准溶液所引入的不确定度urel(ρ)公式(7) 。
(7)
2.2.2 样品称量引入的不确定度 本研究中,两种检测方法称样时所用天平相同,均为精度为0.01g电子天平,样品均匀性及称量重复性引入不确定度均可忽略不计。该级别天平在量程内允许的最大误差为±0.01g,则由称量误差引入的不确定度服从矩形分布(B类评定),两种方法称样量有所不同,则GB/T 20769-2008中由称量引入不确定度urel(mⅠ)计算公式(8),GB 23200.121-2021中由称量引入不确定度urel(mⅡ)计算公式(9)。
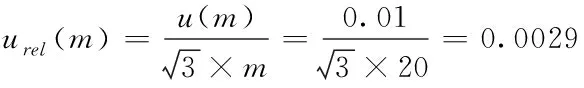
(8)
(9)
2.2.3 样品前处理引入的不确定度 前处理所引入的不确定度主要来源于添加提取试剂、移取及净化后定容时的玻璃器皿的允差及温度变化引起试剂膨胀,均为B类评定。
2.2.3.1 GB/T 20769-2008前处理引入的不确定度 由1.3.1可知,对于GB/T 20769-2008来说,可以引入不确定度的步骤所用器皿包括:添加40mL乙腈所用50mL量筒,允许误差为±0.50mL,读数重复性偏差为±0.10mL;移取提取液所用10mL单标线吸量管,A级允许误差为±0.020mL,读数重复性偏差可以忽略;最终定容所用5mL单标线吸量管,A级允许误差为±0.015mL,读数重复性偏差可以忽略,均服从三角分布。应用公式(4、5)可计算在前处理过程中每次使用玻璃仪器由误差、偏差及温度引入的不确定度(表3)。
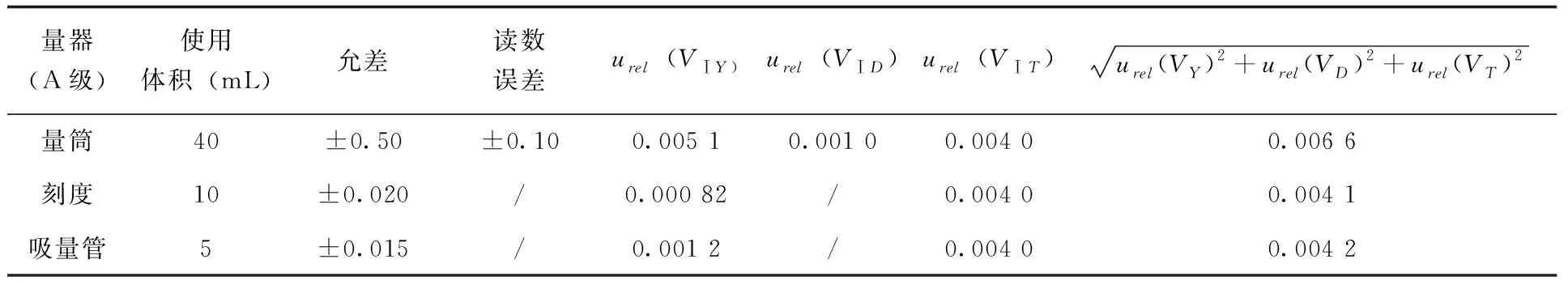
表3 GB/T 20769-2008前处理过程玻璃仪器使用引入不确定度
将上述不确定合并,得到前处理过程引入的不确定度urel(VⅠ)公式(10)。
(10)
2.2.3.2 GB 23200.121-2021前处理引入的不确定度 由1.3.2可知,对于GB 23200.121-2021来说,可以引入不确定度的步骤所用器皿只有在加入10mL乙腈时所用规格为10mL的量筒,其允许误差为±0.10mL,读数重复性偏差为±0.02mL;虽然在后续步骤中有提取液的取用,但这一步骤并不需精确量取,且不参与最终结果的计算,由此可以计算前处理过程引入的不确定度urel(VⅡ)公式(11)。
(11)
2.2.4 仪器检测引入的不确定度 由2.1可知,仪器检测引入的不确定度可以分为标准溶液峰面积引入的不确定度和样品溶液峰面积引入的不确定度。由于超高效液相色谱-串联质谱仪灵敏度较高,可以认为仪器稳定性即测定值的分散性就是构成测量峰面积的不确定度的主要来源,其他因素可以忽略不计。此类不确定度可以在重复性条件下对同一被测量对象独立重复观测多次,最后采用统计方法进行计算,即进行A类评定。在重复性条件下,对标准溶液和样品进行分别进样6次,得到6次进样的峰面积,根据贝塞尔公式(12)计算标准偏差,标准不确定度根据公式(13)计算。
(12)
(13)
根据方法(1、2)的标样溶液和样品峰面积检测结果,分别计算得到相应的标准不确定度及相对标准不确定度(表4)。
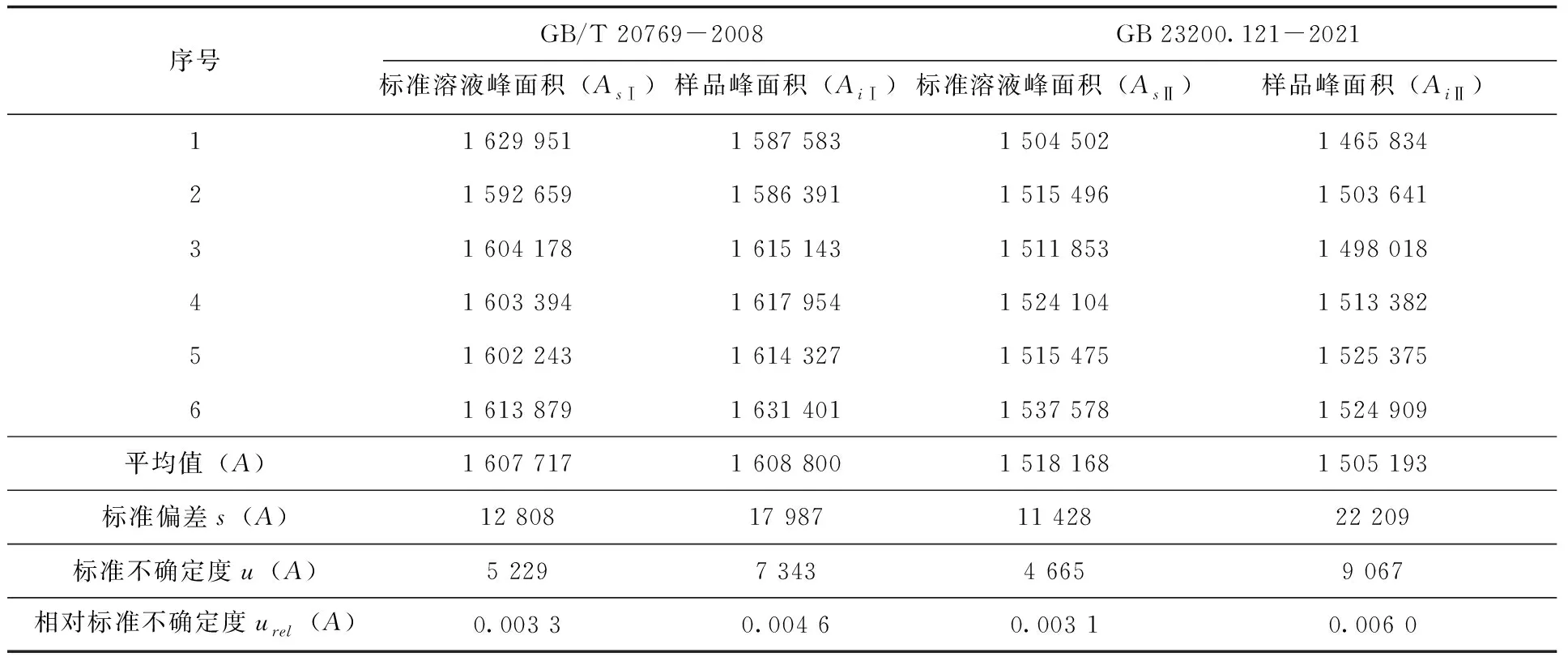
表4 标准溶液和样品峰面积检测结果及不确定度(n=6)
2.2.5 方法精密度引入的不确定度 由方法精密度引入的不确定度符合A类评定,本研究通过检测多个空本样品添加标准溶液进行评定。按照GB/T 20769-2008及GB 23200.121-2021的称样量分别制备5个添加样品并在重复性条件下进行检测,添加浓度均为0.2mg/kg,根据检测结果应用公式(12、13)计算相应标准偏差及标准不确定度,最终计算得到相对标准不确定度(表5)。
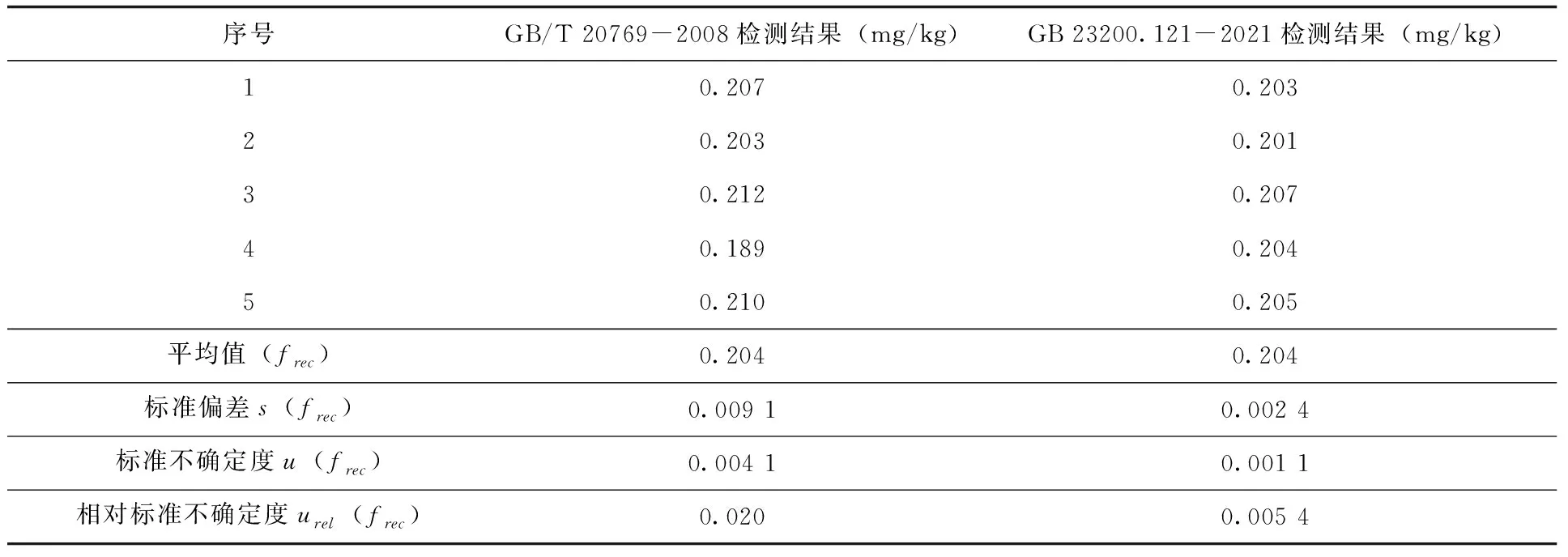
表5 添加回收样品检测结果及不确定度(n=5)
2.2.6 相对标准不确定度的合成 综合上述得到的各不确定度分量,通过公式(3)可计算得到两个方法的合成相对标准不确定度urel(ω),具体各分量不确定度及合成相对不确定度计算结果(表6)。
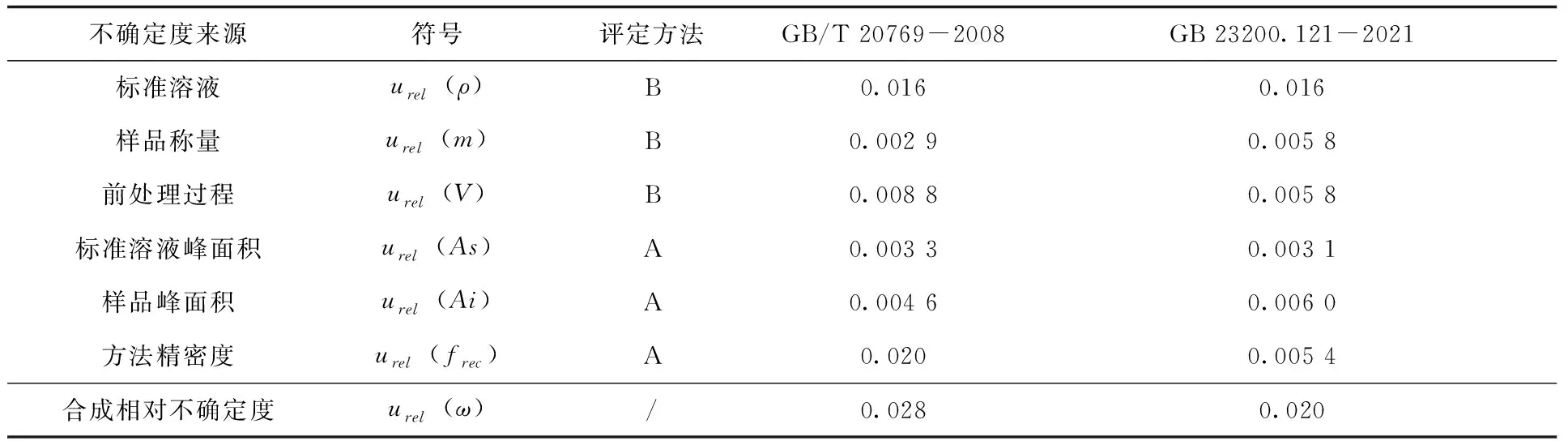
表6 各分量不确定度及合成相对不确定度
由结果可知,对于本次实验来说,两种方法的合成相对标准不确定度均比较低,但依旧存在一定的差异,GB/T 20769-2008明显高于GB 23200.121-2021,主要差别出现在方法精密度上,其他分量引入的不确定度相差并不多。分析原因应该是由于GB/T 20769-2008的前处理步骤相对复杂,需要经过浓缩—净化—浓缩—定容等一系列操作,造成方法精密度较方法二更低,从而提高了不确定度。
对于每个分量对合成相对标准不确定度的贡献度而言,GB/T 20769-2008结果中,贡献度最高的是方法精密度(51.7%)和标准溶液(33.1%);其他分量的贡献度都较低;GB 23200.121-2021结果中贡献度最高的是标准溶液(64.3%),其他各分项贡献度相对较低且比较平均。由此可见,对于两种检测方法来说,标准溶液的不确定度都是影响检测结果不确定度的关键因素,而对于GB/T 20769-2008来说,精密度则是另一个关键因素。
虽然从本次实验的评定结果来看,在不确定度的表现上,GB 23200.121-2021确实优于GB/T 20769-2008,但这一结果仅是反应吡虫啉一种农药在大白菜这一种基质中的情况,并不能得到标准所涉及的所有农药在各种基质中均符合这一规律的结论。这是因为,两种检测方法样品的最终净化效果是差别较大的,GB/T 20769-2008对样品的净化效果要明显优于GB 23200.121-2021,在这种情况下,对于复杂基质,比如含色素较多的绿色蔬菜,含其他杂质较多的鳞茎类蔬菜等,GB 23200.121-2021是否还能保持优于GB/T 20769-2008的精密度,进样溶液杂质的增多是否会影响仪器检测峰面积的重复性等,这些因素都会影响最终不确定度的评定结果,还需要通过进一步的实验具体分析。
2.3 检测结果扩展不确定度的表示 根据测量不确定度评定指南对一般实验室的要求,在置信概率p=95%时,取测量结果的扩展不确定度包含因子k=2,由此得到,对于一个吡虫啉在大白菜样品中残留量的检测结果为MRL值[12](0.2mg/kg)附近的样品来说,用标准方法GB/T 20769-2008和GB 23200.121-2021分别进行检测,其结果的相对扩展不确定度分别为5.6%和4.0%。
3 结论
本研究评定了使用两种液相色谱-串联质谱法检测吡虫啉在大白菜中残留量时结果的不确定度。通过实验及计算可知,由于检测方法更加简便易行,方法重现性更好,新检测标准GB 23200.121-2021的结果不确定度要小于原方法GB/T 20769-2008。但是,本研究只针对吡虫啉一种农药在大白菜基质中,对于更多种农药在其他基质尤其是复杂基质中的不确定度是否符合这一规律还有待更进一步的研究。
猜你喜欢
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
昆钢科技(2021年2期)2021-07-22
科技创新导报(2020年5期)2020-06-11
科教导刊(2017年26期)2017-11-07
考试周刊(2016年94期)2016-12-12
科技与创新(2015年17期)2015-09-11
科技与创新(2014年12期)2014-08-28
中国信息化·学术版(2013年3期)2013-06-25
