
云木香中1个新愈创木烷型倍半萜内酯
2021-07-21 02:27胡祖艳刀建华
中草药 2021年14期
胡祖艳,刀建华,赵 旻
1.西双版纳州食品药品检验所,云南 景洪 666100
2.四川大学华西医院,四川 成都 610041
云木香为菊科(Asteraceae)风毛菊属SaussureaDC.植物云木香Saussrurea costus(Falc.) Lipsch.的干燥根,为多年生草本植物,又名青木香、广木香、兴瞿草[1]。始载于《神农本草经》,列为上品,木香药用历史悠久,干燥根入药,有健脾消食、行气止痛、止痛、安胎的功效[1-2]。现代药理学研究证实,云木香对人体和动物的消化系统[3-6]、呼吸系统[7]、心血管系统[8-9]等方面疾病表现出很好的疗效。云木香的化学成分多样,目前主要集中在挥发油成分的研究,为了深入研究云木香的化学成分,本实验对云木香干燥根的乙醇提取浸膏石油醚萃取部分进行分离纯化,从中分离得到1个新的愈创木烷型倍半萜类衍生物,然后运用高分辨质谱、核磁共振等波谱技术,并结合弱碱水解的方法对其进行结构确证,最后鉴定为云木香素A(costusin A,1),它的2个水解产物为3-epizaluzanin C(1a)和正癸酸(1b)。结构见图1。采用Griess法初步评价化合物1化合物对脂多糖(lipopolysaccharide,LPS)诱导小鼠巨噬细胞RAW 264.7产生NO的抑制活性。结果表明,化合物1对LPS诱导RAW264.7细胞产生NO显示出中等的抑制作用,半数抑制浓度(IC50)为21.7 μmol/L。

图1 化合物1的化学结构Fig.1 Structure of compound 1
1 仪器与材料
Perkin Elmer Model 341型旋光仪(美国PE公司);Perkin Elmer one FT-IR型红外光谱计(美国PE公司);Shimadazu UV-2401紫外可见分光光度计(日本Shimadazu公司);Bruker Ascend 400型核磁共振仪、Bruker MicrOTOF QII型高分辨率质谱仪(德国Bruker公司);LC-3000型高效液相色谱仪(北京创新恒通公司);Kromasil C18反相半制备色谱柱(250 mm×10 mm,5 μm);薄层色谱硅胶GF254和柱色谱硅胶(中国青岛海洋化工厂);50 μm ODS反相C18填料(日本YMC公司);50 μm MCI树脂(日本Mitsubishi Chemical公司)。氘代试剂(美国Sigma Aldrich公司);色谱纯甲醇(天津康科德科技有限公司);其他分析纯化学试剂(天津富宇精细化工有限公司);Griess试剂法NO试剂盒(碧云天生物技术研究所);脂多糖(美国Sigma Aldrich公司);RPMI 1640培养基(美国Gibco公司)。
云木香药材为2019年10月购于成都市荷花池中药市场,由成都中医药大学郭大乐副教授鉴定为菊科风毛菊属植物云木香S.costus(Falc.) Lipsch.的干燥根,样本(HX20191002)存放于四川大学华西医院。
2 提取与分离
云木香干燥根4 kg,粉碎后用95%乙醇常温浸泡提取3次,每次浸提3 d。合并提取液并减压浓缩至浸膏,得总提取物860 g。将总浸膏分散于5 L热水中(60 ℃),用等体积的石油醚萃取3次,减压浓缩得到石油醚浸膏(116 g)。将该石油醚浸膏进行MCI柱色谱除色素,以甲醇-水(9∶1)进行洗脱,得到浸膏98 g。取该石油醚部位进行正相硅胶柱色谱,以石油醚-醋酸乙酯(100∶1→50∶1→20∶1→10∶1→5∶1→2∶1→1∶1)梯度洗脱,经薄层色谱检识合并得到6个组分Fr.A~B(6.2 g)采用ODS中低压柱色谱分离,甲醇∶水(1∶1→3∶2→7∶3→4∶1→9∶1)和甲醇梯度洗脱得到6个组分Fr.B1~B6。Fr.B4(615 mg)经半制备HPLC色谱(78%乙腈)等度分离,得到化合物1(13 mg,tR=31 min)。
3 结构鉴定
化合物1:白色粉末状固体,[α]20D-45° (c0.2,CHCl3)。HR-ESI-MS显示其准分子离子峰为m/z:423.250 9 [M+Na]+(计算值为423.251 1,C25H36O4Na),由此推测其分子式为C25H36O4。红外光谱显示其结构中含有羰基(1 770.97, 1 732.96 cm-1)等基团。
1H-NMR谱(表1)显示36个氢信号,包括6个烯氢信号δH6.26 (1H, d,J= 3.5 Hz), 5.48 (1H, s),5.45 (1H, d,J= 3.1 Hz), 5.33 (1H, s), 4.89 (1H, s) 和4.76 (1H, s);5个次甲基的氢信号δH5.63 (1H, t,J=6.2 Hz), 3.85 (1H, t,J= 9.1 Hz), 3.01 (2H, m), 2.83(1H, m);以及11组亚甲基的氢信号δH2.25 (2H, t,J= 7.5 Hz), 2.08 (1H, m)/2.47 (1H, m), 1.86 (1H,m)/2.24 (1H, m), 1.57 (2H, m), 1.34 (1H, m)/2.20 (1H,m) 和1.20~1.28 (12H, m);另外,高场区有1个甲基氢信号 [δH0.81 (3H, t,J= 6.8 Hz)]。13C-NMR谱(表1)中有25个碳信号,结合HMQC谱分析分别归属为2个羰基碳信号δC173.4, 169.9;6个烯碳信号δC149.3, 148.1, 139.3, 120.4, 113.3, 115.2;5个次甲基碳信号δC85.0, 75.8, 49.8, 45.2, 44.6,其中存在2个连氧的次甲基碳信号δC85.0, 75.8;11个亚甲基碳信号δC37.3, 36.7, 34.5, 31.8, 31.0, 29.4, 29.3,29.2, 29.0, 24.9, 22.6;以及1个甲基碳信号δC14.0。经过文献检索,发现其中主要一维谱信号与愈创木烷型倍半萜3-epizaluzanin C[10]的信号非常相似,剩余信号又与正癸酸标准物质信号[11]相吻合。因此,初步判断此化合物的结构特征为愈创木烷型倍半萜母核上连有长链饱和脂肪酸。

表1 化合物1的1H-NMR (400 MHz, CDCl3) 和13C-NMR (100 MHz, CDCl3) 数据Table 1 1H-NMR (400 MHz, CDCl3) and 13C-NMR (100 MHz, CDCl3) data for compound 1
然后,通过二维谱对其平面结构进行进一步的鉴定(图2)。在HMBC谱中,H-1 (δH3.01) 与C-4(δC149.3), C-14 (δC113.3), C-6 (δC85.0), C-3 (δC75.8) 存在远程相关;H-2 (δH1.86, 2.24) 与C-10 (δC148.1), C-5 (δC44.6) 有远程相关;H-5 (δH3.01) 与C-10 (δC148.1), C-7 (δC45.2), C-2 (δC37.3) 存在远程相关;H-6 (δH3.85) 与C-4 (δC149.3), C-1 (δC49.8), C-8 (δC31.0) 有远程相关;另外,结合1H-1H COSY谱中H-3/H-2/H-1/H-5/H-6/H-7/H-8/H-9相邻之间给出的相关信号;证明了其中五元环与七元环是通过1,5位碳进行骈合。H-7 (δH2.83) 与C-12 (δC169.9), C-13 (δC120.4), C-5 (δC44.6), C-9 (δC36.7)存在远程相关;H-13 (δH5.45, 6.26) 与C-11 (δC139.3), C-12 (δC169.9), C-7 (δC36.7) 有远程相关,可以推测出异丙烯酸连接在C-7位上并与C-6位羟基形成内酯。另外,连在C-4位的双键位置可以根据H-15 (δH5.33, 5.48) 与C-4 (δC149.3) 和C-3 (δC75.8) 的远程相关信号确定,而连在C-10位的双键位置由H-14 (δH4.76, 4.89) 与C-10 (δC148.1), C-1(δC49.8), C-9 (δC36.7) 的远程相关信号确定。最后,根据H-3 (δH5.63) 与C-1′ (δC173.4) 的远程相关信号可以确定脂肪酸通过与C-3位羟基形成酯键相连。综上,化合物1的平面结构鉴定为3-epizaluzanin C-3-yl decanoate(图2)。另外,H-3与H-6的相对构型可以通过NOESY谱中H-3 (δH5.63) 与H-6 (δH3.85) 的相关信号得到判断(图3)。

图2 化合物1的主要HMBC和1H-1H COSY相关Fig.2 Key HMBC and 1H-1H COSY correlations of compound 1
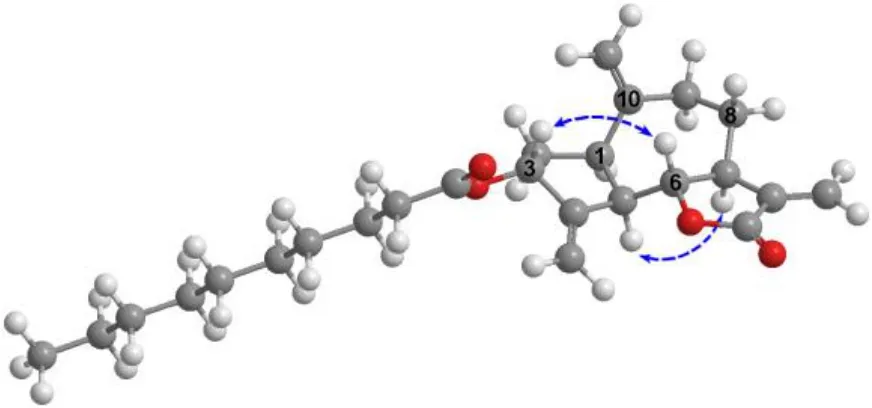
图3 化合物1的主要NOESY相关Fig.3 Key NOESY correlations of compound 1
为了对化合物1进行完整的结构确证,用0.1 mol/L KOH在常温中对其进行温和的碱水解,反应完成后加酸中和,用CH2Cl2萃取,萃取产物经HPLC制备得到水解产物1a和1b。通过与已知化合物3-epizaluzanin C[10]和正癸酸[11]进行波谱数据及理化性质的比对,确定化合物1a和1b分别为3-epizaluzanin C和正癸酸。综上所述,化合物1的结构鉴定如图1所示,经Scifinder检索确定为新的愈创木烷型倍半萜衍生物,命名为云木香素A。
化合物1a:白色粉末状固体,[α]20D-45° (c0.2,CHCl3),HR-ESI-MS给出准分子离子峰m/z269.115 2[M+Na]+(计算值为269.115 4),确定分子式为C15H18O3。1H-NMR (400 MHz, CDCl3)δ: 6.18 (1H, d,J= 3.5 Hz, H-13β), 5.47 (1H, d,J= 3.1 Hz, H-13α),5.45 (1H, s, H-15β), 5.32 (1H, s, H-15α), 4.90 (1H, s,H-14β), 5.75 (1H, s, H-14α), 4.65 (1H, t,J= 6.2 Hz,H-3), 3.87 (1H, t,J= 9.2 Hz, H-6), 3.07 (2H, m, H-1,5), 2.83 (1H, m, H-7), 2.49 (1H, m, H-9β), 2.21 (1H,m, H-8β), 2.14 (1H, m, H-2β), 2.08 (1H, m, H-8α),1.84 (1H, m, H-2α), 1.36 (1H, m, H-9α);13C-NMR(100 MHz, CDCl3)δ: 49.6 (C-1), 36.9 (C-2), 74.4(C-3), 154.1 (C-4), 45.6 (C-5), 85.2 (C-6), 44.1 (C-7),31.1 (C-8), 39.9 (C-9), 139.5 (C-10), 148.6 (C-11),170.2 (C-12), 120.5 (C-13), 113.2 (C-14), 112.9(C-15)。以上数据与文献报道一致[10],故鉴定化合物1a为3-epizaluzanin C。
化合物1b:白色结晶状固体,HR-ESI-MS给出准分子离子峰m/z172.146 3 [M+Na]+(计算值为172.146 3),确定分子式为C10H20O2。1H-NMR (400 MHz, CDCl3)δ: 2.34 (2H, t,J= 7.5 Hz, H-2), 1.63(2H, m, H-3), 1.35~1.27 (12H, m, H-4~9), 0.88 (3H,t,J= 6.8 Hz, H-10);13C-NMR (100 MHz, CDCl3)δ:180.6 (C-1), 34.3 (C-2), 24.8 (C-3), 29.2 (C-4), 29.4(C-5), 29.5 (C-6), 29.4 (C-7), 32.0 (C-8), 22.8 (C-9),14.2 (C-10)。以上数据与文献报道一致[11],故鉴定化合物1b为正癸酸。
4 NO抑制活性测定
参考文献采用Griess法[12]评价化合物1对RAW 264.7细胞NO释放的抑制作用。取处于对数生长期的RAW 264.7细胞,按1×105个/mL浓度进行稀释,稀释液加如96孔培养板中,每孔加入200 μL细胞悬液。随后,在37 ℃、5% CO2培养箱中培养1 h,每孔再加入LPS(1 μg/mL)及不同浓度(5.0、10.0、20.0、40.0 μmol/L)的测试样品,然后在37 ℃、5% CO2培养箱中培养24 h后吸取培养液上清液100 μL转移到酶标板中,在540 nm下测定吸光度(A),结果以IC50表示。结果显示,化合物1对LPS诱导RAW264.7细胞释放NO显示出一定的抑制活性,IC50为21.7 μmol/L,而阳性药吲哚美辛的IC50为15.4 μmol/L。
5 讨论
本实验采用各种色谱分离技术与波谱手段,并结合化学方法,从云木香中分离鉴定1个新愈创木烷型倍半萜,命名为云木香素A,该化合物对LPS诱导RAW 264.7细胞释放NO显示出抑制作用,可为云木香的深入开发与利用提供参考。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
烟草科技(2022年6期)2022-06-27
重庆大学学报(2022年5期)2022-06-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中成药(2020年2期)2020-05-12
天然产物研究与开发(2019年1期)2019-03-01
中成药(2017年10期)2017-11-16
中成药(2017年5期)2017-06-13
光谱学与光谱分析(2016年6期)2016-07-12
——青蒿素
中国学术期刊文摘(2015年21期)2015-12-24
中国药业(2014年21期)2014-05-26
