
Targeting EZH2 for the treatment of soft tissue sarcomas
2021-07-20 03:13MagdalenaKarolakIanTracyJanetShipleyZoWalters
Magdalena Karolak, Ian Tracy, Janet Shipley, Zoë S Walters
1Translational Epigenomics Team, Cancer Sciences, Faculty of Medicine, University of Southampton, Southampton SO16 6YD,UK.
2Sarcoma Molecular Pathology Team, Divisions of Molecular Pathology and Cancer Therapeutics, The Institute of Cancer Research, London SM2 5NG, UK.
Abstract Soft tissue sarcomas (STS) are a heterogenous group of rare malignancies of mesenchymal origin, affecting both children and adults.The majority of STS have a poor prognosis and advanced stage at the time of diagnosis.Standard treatments for STS largely constitute tumour resection with chemotherapy and/or radiotherapy, and there has been little significant advancement in the application of novel therapies for treatment of these tumours.The current multimodal approach to therapy often leads to long-term side effects, and for some patients,resistance to cytotoxic agents is associated with local recurrence and/or metastasis.There is, therefore, a need for novel therapeutic strategies for the treatment of STS.Recent advances in epigenetics have implicated the histone methyltransferase, EZH2, in the development and progression of diseases such as breast cancer, lymphoma and more recently STS.Here we will review the current literature for EZH2 in STS, including high expression of EZH2 in STS and correlation of this with specific features of malignancy (metastasis, histological grade, and prognosis).The effects of targeting EZH2 using RNA interference and small molecule inhibitors will also be reviewed and the potential for the use of EZH2 inhibition in therapeutic strategies for STS patients will be discussed.
Keywords: EZH2, epigenetics, therapy
INTRODUCTION
Soft tissue sarcomas (STS) constitute a highly heterogenous group of rare malignancies believed to be ofmesenchymal origin.Mesenchymal stem cells harbour powerful self-renewal capacity and multi-lineage differentiation potential into various tissues, such as adipose, muscle, bone and cartilage.Over 60 distinct STS subtypes have been identified with regard to their biological features and clinical manifestation, making the classification of these tumours often problematic and challenging.STS can occur in any part of the body,but the most frequent sites include the extremities, head and neck areas as well as trunk.STS comprise around 1% of all adult cancers and around 8% of all paediatric and adolescent tumours.However, the spectrum of STS subtypes differs in these different age ranges.Despite being relatively rare cancers, the vast majority of diagnosed cases of STS present with highly aggressive behaviour, poor prognosis and advanced stage of the disease at point of detection.Many STS can be thought of as undifferentiated tumours, where STS cells express early markers of lineage-specific differentiation but do not differentiate into the mature,benign tissue type.For example, rhabdomyosarcomas (RMS) resemble immature skeletal muscle cells that have failed to complete differentiation and cell cycle arrest, and the restoration of this process is considered to be of therapeutic value[1,2].
A number of STS discussed in this review, harbour specific genetic hallmarks, such as chromosome translocations and specific gene mutations or amplification events.These contribute to the process of oncogenesis and may be useful diagnostic features.An example is the recurrent chromosomal translocation t (X;18) (p11.2;q11.12) found in synovial sarcomas (SS), leading to gene fusions betweenSS18on chromosome 18 and eitherSSX1,SSX2or in rare casesSSX4on chromosome X.The abnormal fusion protein SS18-SSX disrupts the epigenetic regulation of gene expression and is believed to drive sarcoma formation in SS[3,4].Another example is malignant peripheral nerve sheath tumours (MPNSTs), in whichNeurofibromin(NF1) tumour suppressor gene mutations are thought to drive malignancy in some patients.Such mutations lead to inactivation of NF1 protein and therefore to development and pathogenesis of MPNSTs[5].The same holds true for extremely rare and aggressive atypical teratoid rhabdoid tumours(ATRTs, brain) or malignant rhabdoid tumours (MRTs, kidneys and soft tissues) where homozygous inactivation ofSMARCB1, and resultant deficiency of SMARCB1 protein, a member of the SWI/SNF complex, occur in the majority of these malignancies[6].RMS, ATRT and MRT tumours affect predominantly infants and/or children/adolescents, with RMS being the most frequent, accounting for 50%of all STS in childhood[2,7].
Current treatment of STS is largely based on tumour resection followed by chemotherapy and/or radiotherapy.This multimodal approach often leads to long-term side effects and, due to resistance to cytotoxic agents in large proportion of sarcoma patients, it also results in local recurrence as well as metastasis.Despite intensification of treatment regimes, little significant advancement in treatment outcomes for high-risk patients with these malignancies has been noted in recent years[8].The low efficiency/failure of the standard therapies in STS highlights a fundamental necessity for development of novel, more effective and less harmful treatment strategies for sarcoma patients.
ROLE OF EZH2 IN STS
Enhancer of Zeste Homologue 2 (EZH2) is the catalytic subunit of Polycomb Repressive Complex 2(PRC2), a multiprotein complex comprised of four core units, EED, SUZ12, RbBP4, and EZH2, although a number of other auxiliary proteins have also been shown to modulate PRC2 activity [Figure 1][9].PRC2 is crucial for maintaining the epigenetic state of the cells through modulation of chromatin structure and by such means, regulation of gene expression.EZH2 is a histone methyltransferase whose mode of action is observed as addition of methyl groups at lysine 27 in histone H3 through its active SET domain motif,resulting in the formation of the H3K27me3 epigenetic mark and subsequent transcriptional repression[10].EZH2/PRC2-facilitated methylation of H3K27 is part of a gene expression regulatory process, and the endeffect of this chain of events is the compaction of chromatin from “open” euchromatin to “closed”heterochromatin and functional repression of gene expression by preventing the binding of nuclear transcription factors [Figure 2].Many of the genes which EZH2 is involved in silencing are effectors of cellular differentiation[11].Thus, EZH2 activity is broadly associated with the prevention of terminal differentiation and lineage commitment in cells, and maintaining the capacity for self-renewal and a pluripotent stem cell phenotype in those cells with its high expression[12].Through its role in gene expression regulation, EZH2 is involved in a variety of biological processes, including regulation of cell cycle, cell differentiation, cell proliferation, division, and senescence.
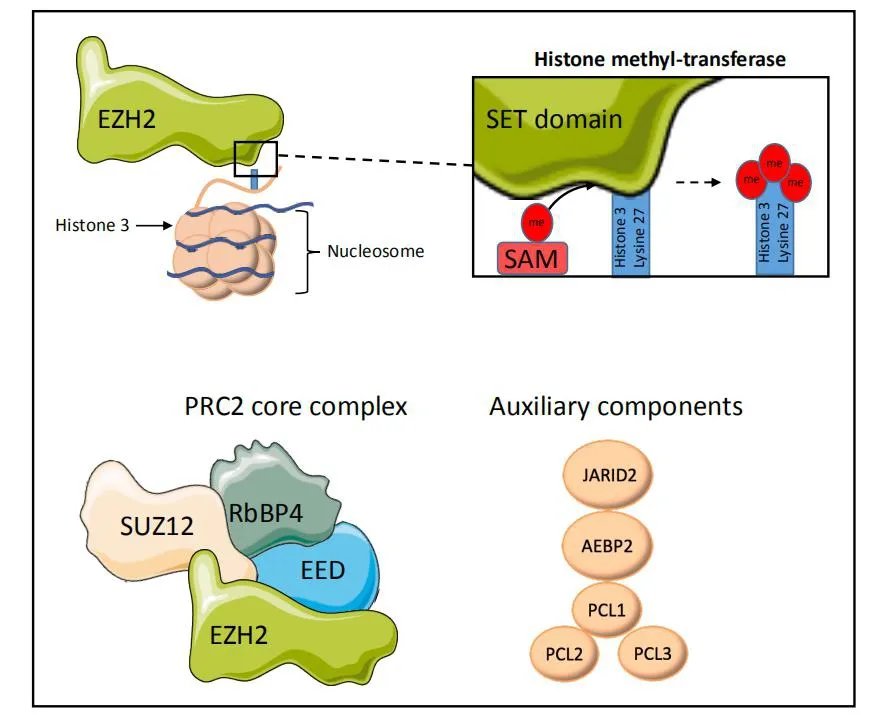
Figure 1.EZH2 is a histone methyltransferase, catalysing the transfer of donor methyl groups from SAM to H3K27 via its C-terminal SET domain.EZH2 predominantly functions as part of a multi-protein complex, PRC2, containing the core units SUZ12, EED, and RbBP4,which aid in recognition and targeting of EZH2 activity to histone 3 lysine-27.A number of other auxiliary proteins are known to complex with PRC2 but are disposable to its core activity.All structures are representative only.Subset of images from Servier medical art https://smart.servier.com/?s=nucleosome.SAM: S-adenosyl-L-methione.
A recent meta-analysis of 8 studies has shown that mutations inEZH2confer a poorer prognosis for patients with myeloid neoplasms, such as acute myeloid leukaemia[13].Likewise results from a Phase-II trial of the EZH2 inhibitor, Tazemetostat, in relapsed or refractory follicular lymphoma patients withEZH2mutations have promising initial results[14].However, mutations ofEZH2are not prevalent in soft tissue sarcomas.A study in 2012 screened a range of sarcoma types for mutations in a selection of seven genes,
includingEZH2, known to be recurrently mutated in non-sarcomatous cancers.Of these genes only twoPIK3CAmutations and oneJAKmutation were observed in the entire cohort of samples (3/108; 2.8%).The soft tissue sarcomas investigated in that study included malignant fibrous histiocytomas,rhabdomyosarcomas, malignant peripheral nerve sheath tumours, leiomyosarcomas, synovial sarcomas,liposarcomas, angiosarcomas, and Ewing sarcomas[15].
Rather, EZH2 is overexpressed in many STS in comparison to normal tissue, which may be attributed to the cell of origin of these tumours.STS are thought to be derived from mesenchymal stem cells, where the balance of EZH2 expression along with other histone modifying enzymes is thought to control the differentiation lineage process[16-19].The reason why overexpression of wildtype EZH2 leads to associated poor outcomes can primarily be understood in terms of the normal function of EZH2.EZH2 is involved in silencing genes which drive differentiation in developing cells/tissues[20].Under normal development, EZH2 expression fades over time, to become almost undetectable in adult specialized cells and tissues [Figure 3][10].The inappropriately timed expression of EZH2 is therefore thought to lead to an undifferentiated phenotype with cells maintaining the ability for self-renewal.
Of increasing importance and focus are interactions between PRC2 and SWItch/Sucrose Non-Fermentable(SWI/SNF) complex.The latter is another chromatin remodelling machinery with an opposed function to PRC2[10].The antagonistic mode of action of both complexes is essential to ensuring homeostasis and proper epigenetic profile of the cells.Mutations in the SWI/SNF complex subunitSMARCB1(also referred to asINI1) result in loss of SMARCB1 regulatory function, leading to the dysregulation of EZH2 activity and thus abnormal gene repression and oncogenic capacity of EZH2[21-23].As a consequence, it is observed as development and progression of solid tumours[24].Nonetheless, the EZH2 aberration linked to SWI/SNF constitutes only one out of many other causes of EZH2 dysregulation and subsequent pathogenesis[25].
CORRELATIONS OF EZH2 WITH CLINICAL FEATURES
EZH2 upregulation
Higher levels of EZH2 have been noted in MPNST tumours and MPNST cell lines, compared to normal human Schwann cells[5].The same phenomena were detected in RMS tumour samples and cell lines, with little or noEZH2expression observed in normal skeletal muscle tissue[26], benign rhabdomyoma or tumouradjacent skeletal muscle[20].This is also true for leiomyosarcoma, where EZH2 exhibited elevated levels with high sensitivity and specificity in malignant tissues in contrast to benign leiomyoma or normal myometrium[20].Another study investigating the expression ofEZH2in embryonal (ERMS) and alveolar(ARMS) RMS cell lines and patient samples showed similar findings demonstrating high upregulation of the RNA in comparison to myoblasts as a control[27].
Prognosis
In STS prognosis is usually poor, dependent on other factors such as stage of malignancy and tumour size at time of detection, the patient’s age, localisation of the tumour and presence/ absence of metastatic lesions.In the prognosis of SS, high levels of EZH2 protein are associated with worse clinical outcome, especially in the poorly differentiated subtype[28].In a study including 104 patient-derived tumour samples (with 27 having prior chemotherapy, 25 radiotherapy and 13 both) of mixed STS: SS, leiomyosarcomas, RMS,epithelioid sarcomas, malignant fibrous histiocytomas, myxofibrosarcomas and liposarcomas, higher levelsof EZH2 protein were independently correlated with unfavourable prognosis for each STS type[29].
Metastasis
In studies comprising 55 SS primary tumour samples without preoperative chemo- or radiotherapy[28], and 29 SS tumour samples (22/29 treated with chemotherapy and radiation following surgery)[30], higher levels of EZH2 and H3K27me3 as examined by immunohistochemistry (IHC) were observed in patients where distant metastasis has occurred[28,30].EZH2 was also significantly associated with distant metastasis in other sarcoma subtypes, such as LMS, ES, LS, RMS and MFH[29].In a study conducted on 17 paediatric RMS and extraosseous Ewing sarcoma (EES) tumour samples, elevated EZH2 protein levels were associated with increased aggressiveness of the disease and the presence of metastasis to lymph nodes and/or distant parts of the body at the time of diagnosis[31].
Survival
Higher EZH2 expression in 14 patients with SS was significantly associated with worse survival.However,the same was not found to be significant in 31 liposarcoma and 36 MFH patients[29].In a study conducted on 105 sarcoma tumour samples from patients with no prior adjuvant therapy (28 RMS, 15 Ewing’s Sarcoma,30 osteosarcoma, 18 SS, 14 epithelioid sarcoma and chondrosarcoma), higher EZH2 expression and higher H3K27me3 levels were associated with significantly shortened overall survival[32].However, the inclusion of tumour types other than STS means that this may therefore not represent the true OS for these types of malignancy.Expression of EZH2 was also a marker of lower likelihood of survival in paediatric samples of 11 RMS and 6 EES tumours, compared to the EZH2-negative cases[31].Furthermore, 14 out of 29 SS patients with lower EZH2 levels, had a significantly prolonged overall survival than those of higher EZH2 expression profiles[30].
Histological grade and clinical stage
In a study using 104 patient-derived tumour samples, it was observed that higher EZH2 expression was associated with higher histological grade of the tumour[29].This is inconsistent with another study, in which EZH2 high expression did not show significant correlation with respect to histological grade[30].This may be due to the smaller sample size (29 SS cases).In another paper, consisting of 50 patients-derived SS tumour samples, those with II and III clinical stages had significantly higher EZH2 scores by immunohistochemistry(IHC) than those with stage I with no EZH2 expression found in one case with stage IV[3].In 17 patients with RMS and EES, high intra-tumour EZH2 scores were present in stage III or IV of the malignancy[31].These inequalities may emerge due to the limited sample sizes, so further investigations are needed.
EZH2 and Ki-67
In SS EZH2 expression was correlated with Ki-67 scores, especially in poorly differentiated subtypes[28].Abundant expression of Ki-67 with strong positive correlation to EZH2 scores was also observed in other types of STS, such as LMS, alveolar and embryonal RMS[33], LS, ES and MFH[29].These clearly indicated the positive association between EZH2 expression and proliferative potential in soft tissue malignancies.In another study including various subtypes of RMS, such as ARMS, ERMS, unclassified, spindle cell and pleomorphic RMS, the Ki-67 and EZH2 were also parallel in all primary and recurrent RMS tumour samples[34].
TARGETING EZH2 IN STS
EZH2 modulation through RNA interference
Rhabdomyosarcoma
Knockdown ofEZH2through vector-based shRNAs in the embryonal RMS cell line RD resulted in induction of differentiation due to the increase of muscle specific factors, restored recruitment of multiprotein complexes at muscle-specific promoters and activated expression of MyoD, as well as its binding abilities, crucial for myogenesis[1].Another study showed that depletion ofEZH2with siRNAs in the RD cell line led to inhibition of cell proliferation and significant decrease of H3K27me3.These were followed by the expression of proteins specific to terminal myogenic differentiation[35,36], indicating that the ablation ofEZH2restores myogenic differentiation ability of embryonal RMS cells.Alveolar RMS cell lines(RH30 and RH4, containingPAX3-FOXO1fusion gene), which underwentEZH2interference through siRNA or shRNA manifested inhibition of cellular proliferation, migration, anchorage-independent growth and reduction of cell survival[36-38].Surprisingly, theEZH2depletion led to expression of pro-apoptotic genes instead of differentiation genes, resulting in initiation of cellular death[37].The authors demonstrate that the mechanism of apoptosis in these cells is likely due to derepression of pro-apoptotic gene including the tumour suppressorFBXO32afterEZH2knockdown, as well as in reduction of myogenic proteins such as Myogenin and MyoD, indicating an induction of apoptosis over differentiation[37].A separate study also demonstrated apoptosis in RH30 cells after siRNA silencing of EZH2.However, they also showed that modest reduction ofEZH2in both ERMS (RD) and ARMS (RH30) cells led to differentiation and expression of the terminal differentiation marker myosin heavy chain (MHC) in the presence of differentiation conditions[36].It is worth noting that EZH2 is involved in a feedback loop with other regulatory components such as miR-101, which may serve as a tumour suppressor, and through repression of this molecule is able to promote tumorigenesis in ERMS, as was shown in a study conducted by Vella and colleagues[2].Silencing of EZH2 through siRNAs in ERMS cell lines increased the expression of several miRNAs such as miR-29b, miR-214 and miR-101.The latter caused down-regulation of both mRNA and protein levels of EZH2 and thus, reduction of the tumorigenic potential of ERMS cellsin vitro[2].
Rhabdoid Tumours (MRTs and ATRTs)
Silencing ofEZH2by shRNAs in SMARCB1-deficient paediatric ATRT cell lines (BT12, BT16 and patientderived UPN 737) has been shown to reduce cell proliferation and concomitantly induce cellular senescence and apoptosis, with the biggest effects seen with combination EZH2-silencing by shRNA and administration of DZNep.It was also demonstrated that inhibition of EZH2 affected crucial pathways and molecules involved in cell cycle regulation with downregulation of E2F and c-Myc[39].Furthermore, in an MRT cell line model, sustained knockdown ofEZH2through siRNA manifested as reduction of cellular growth and SMARCB1 restoration[40].
Other STS Subtypes (MPNST, SS, LS)
Stable knockdown ofEZH2using shRNAs in both non-NF1 related and NF-1 related MPNST cell lines resulted in increased apoptosis, inhibited growth and decreased viability of malignant cellsin vitro.By using MPNST immunodeficient mouse xenografts it was demonstrated that injectedEZH2-targeted shRNAs were also able to inhibit tumorigenicityin vivo[5].In the SS cell lines possessing either SS18-SSX1 (Aska-SS,Yamato-SS) or SS18-SSX2 translocation (Fuji, SYO-1) the silencing ofEZH2by siRNA and shRNA led to dose-dependent inhibition of cell proliferation in all four cell lines and decreased levels of H3K27me3[3].Conversely, depletion of EZH2 through RNAi in well-differentiated and dedifferentiated liposarcoma cell lines with addition of steroids and insulin, led to cell proliferation inhibition, followed by induction of differentiationin vitro[41].
EZH2 modulation through use of small molecular inhibitors
Recently, EZH2 has been the focus of numerous drug discovery efforts, resulting in a number of tool compounds and clinical candidates targeting EZH2 being developed.The first to be developed was 3-deazaneplanocin A (DZNep), whose mechanism of action is as an S-adenosyl-L-homocysteine (SAH)hydrolase inhibitor.DZNep represses S-adenosyl-L-methionine-dependent histone methyltransferase activity and thus is not a specific EZH2 inhibitor.A number of S-adenosyl-methionine competitive EZH2-specific inhibitors have since been developed [Table 1], a number of which have been taken into clinical trials for blood cancers and solid tumours (Tazemetostat and GSK126)[14,42-45].

Table 1.Table of the EZH2 inhibitors discussed in the review, along with their specificity over EZH2, effects on the protein and mechanisms of action.
Synovial sarcoma
In SS18-SSX1 (HS-SY-II) and SS18-SSX2 (Fuji) translocation-positive and SMARCB1-deficient synovial sarcoma cell lines, treatment with tazemetostat led to a concentration-dependent inhibition of cell growth and apoptosisin vitro.Administration of tazemetostat or EPZ011989 in xenograft models of SS, carrying either the HS-SY-II/Fuji cell line or patient-derived tumour, resulted in a dose-dependent decrease in tumour volume and inhibition of tumour growth, followed by decreased H3K27me3in vivo.Combined therapy of tazemetostat and the chemotherapeutic drug doxorubicin demonstrated remarkable antitumor activity in Fuji xenografts, relative to each monotherapy alone in the course of treatment.However, after discontinuance of dosing, tumour regrowth was observed.The same treatment regimen in HS-SY-II xenografts did not show the same effects, and instead dose-dependent reduction of intra-tumoral H3K27me3 levels was seen, which was attributed to additional genetic aberrations in this cell line.In patient-derived xenografts, two out of three models exhibited significant inhibition of tumour growth after administration of tazemetostat, most markedly in xenografts with highest SMARCB1 deficiency[4].This suggests the SS18-SSX translocation-positive SS cells are sensitive for treatment with tazemetostat.Dosage of another EZH2 inhibitor, EPZ005687 in SS18-SSX translocation-positive four cell lines (Aska-SS, SYO-1,Fuji, Yamato-SS) resulted in decreased expression of H3K27me3, and dose-dependent inhibition of cell proliferation and migration (up to 48hrs after 72hrs of dosing).However, the combined treatment of EPZ005687 with three chemotherapeutic drugs (etopside/topotecan/doxorubicin) in Aska-SS and SYO-1 cell lines did not show significant synergy[3].In a phase 2 clinical trial involving 33 SS patients with confirmed either SMARCB1 loss/depletion or SS18-SSX translocation and median of 2 preceding systemic treatments, single agent tazemetostat treatment resulted in stable disease in 11 patients with only 5 of those having stable disease lasting 16 weeks or more.No objective responses in pre-treated patients were seen[47].
Rhabdomyosarcoma
A study comprising ERMS RD cell line and administration of a differentiating agent 12-O-tetradecanoylphorbol-13-acetate (TPA) in combination with GSK126 led to markedly more differentiation(assessed by observation of morphological changes in cell phenotype) than the use of either drugs alone,coupled with an increase in MHC expression.Dosage of the drugs after differentiation-induction led to a reduction in cell growth[48].The treatment with DZNep or two EZH2 inhibitors, MC1948 and the more potent MC1945, in the ERMS RD cell line showed dose-dependent inhibition of the RMS cells proliferation and reduction of H3K27me3.Furthermore, immunofluorescence analysis for MHC revealed signs of myotube-like structures, suggesting restoration of myogenic differentiationin vitro.The same treatment regimen with MC1945 applied to mouse xenograft resulted in reduction of tumour growth and induction of differentiationin vivo, which confirms anti-tumour and pro-differentiative activity of these inhibitors in both, cell lines and living organism[35,38].Similarly, administration of either DZNep or MC1945 to ARMS cell lines significantly affected their proliferative potential and led to manifestation of pro-apoptotic cell features, mirroring the RNAi data.The same results were obtainedin vivo, demonstrating reduction of tumour volume and depletion of EZH2 and Ki-67-positive cells in a mouse xenograft[37,38].Notably, EZH2 inhibition in ARMS led to apoptosis, whereas in ERMS resulted in induction of differentiation.The possible interpretation of these may be that both subtypes originate from the same cellular lineage but undergo distinct differentiation pathways and generate specific clinical phenotypes[49], which determines their response to the therapy.
A more recent paper describes a new treatment approach involving dual “hybrid” EZH2/HDAC inhibitor in RH4 cell line.This strategy led to cell cycle arrest, apoptotic events, affected cell viability, enhanced differentiation and reduction of H3K27me3.The higher doses of the dual drug were more efficient and worked in an intensified manner[50], which confirms the more complex or combined treatment in STS at certain conditions works better than monotherapy.Accordingly, the concurrent administration of TPA/GSK126 and Vincristine to chemoresistant and undifferentiated MYOD1+/NOG+ ERMS cells led to enhanced effectiveness of the therapy and increased synergy.The administration of TPA/GSK126 at first showed growing levels of MYOG protein, indicating cell differentiation.Following treatment with vincristine, higher decreases in cell viability and reduction of surviving RMS cells were observed, suggesting drugs synergy[51].Administration of DZNep to ERMS cells led to restoration of several micro-RNAs, such as miR-29b, miR-214 and miR-101.The same results were obtained through forced expression of a mature miR-101 precursor, leading to restoration of miR-101 levels, followed by down-regulation of both mRNA and protein levels of EZH2 and therefore, reduction of the tumorigenic potential of ERMS cellsin vitro.These discoveries confirm the miR-101 is directly targeted by EZH2 and lower levels of this microRNA may contribute to EZH2 overexpression and hence, promote cancer progression in ERMS[2].In contrast to these findings, the Paediatric Preclinical Testing Program (PPTP) study demonstrated that administration of the highly potent and EZH2-selective inhibitor tazemetostat as a single agent in ARMS mouse xenografts did not show promising antitumour activity[52].
Malignant rhabdoid tumours
The effectiveness of tazemetostat has also been tested against rhabdoid tumoursin vivoby the PPTP.Tazemetostat was administered to immunodeficient mouse MRT xenografts, demonstrating significant antitumor activity and differences observed in event free survival (EFS) in 5 out of 7 rhabdoid xenografts,compared to non-rhabdoid models.Both, MRT and ATRT xenografts in which the EZH2 inhibition was the most effective, were identified as SMARCB1-deficient[52].In another study, administration of EPZ-6438 in MRT cells led to a dose-dependent decrease in H3K27me3, with other histone epigenetic marks being unaffected.Furthermore, proliferation of cancerous cells was significantly reduced compared to controlSMARCB1wild-type cells.EPZ-6438 treatment in immunocompromised mouse xenografts bearing G401 cells showed completed elimination of the tumour without recurrence after termination of dosing.Smaller doses of the drug resulted in stable tumour growth at first, followed by delay in growth[6].Another approach evaluating DZNep in combination with conventional cytostatic drugs (doxorubicin/etoposide) or epigenetic agents (5-Aza-CdR/SAHA) to three, anatomically distinct MRT cell lines (kidney, brain, and liver)displayed significantly synergy.Addition of DZNep led to remarkable intensification of anti-proliferative outcomein vitro[7], except for treatment with doxorubicin, in which addition of DZNep did not showsignificant differences[53].In another study, treatment of G401 cells with GSK126, GSK343 or UNC1999 showed reduction of H3K27me3 levels and only modest inhibition of cell proliferation.Furthermore, the combination of a histone deacetylase inhibitor, Panobinostat, with GSK126 resulted in a significant reduction in EZH2 expression, and anti-proliferative and pro-differentiation effectsin vitroandin vivo,compared to use of a single agent[40], as demonstrated in other tumour types[54-57].
Atypical teratoid rhabdoid tumours
In ATRTs, use of DZNep resulted in anti-proliferative and pro-apoptotic effects, increased sensitivity of cancerous cells to radiation and decreased ability to self-renewal and to form tumour spheres[39].Another study showed that cell viability was significantly impairedin vitro, but administration of DZNepin vivodid not change the survival time relative to control or systemic-treated/combination-treated xenografts[58].By contrast, in the PPTP study, administration of tazemetostat to immunodeficient mouse ATRT xenografts led to significant differences in EFS, compared to control[52].This suggests that EZH2-specific inhibition as a single agent is unlikely to be a credible therapeutic option for these patients.Another combination therapy to be tested in ATRT was GSK126 with JQ1 (a bromodomain inhibitor).This resulted in enhanced inhibition of cell proliferation and invasionin vitro, suppressed tumour growth and extended survival in mouse xenografts, compared to the use of each drug alone[59].
Epithelioid sarcoma
A study involving patient-derived INI1-negative ES tumour samples xenotransplanted to immunodeficient mice and administration of EPZ011989 resulted in tumour growth stabilization at first, followed by tumour volume inhibition of up to 89% and decreased H3K27me3[60,61].A clinical trial of patients with SMARCB1(INI1)-negative ES treated with tazemetostat showed similar results to SS patients.Of 62 treated participants, with a median of 1 prior therapy, 15% exhibited partial responses with objective response rate and disease control rate of 15% and 26%, respectively.The duration of response to the treatment ranged from 7.1 to 103.0 weeks and a median overall survival of 82.4 weeks was observed for all 62 patients[60].
Other STS Subtypes (MPNST, LMS, LS)
A study involving NF1-mutant (S462) and non-NF-1 mutated MPNST (MPNST724) cell lines treated with DZNep demonstrated dose-dependent apoptosis of the cellsin vitrorelative to untreated normal human Schwann cells.The viability of the cells, measured by the MTT assay, was significantly reduced from 100%to 30% and 50% in S462 and MPNST724 cell lines, respectively.Cell cycle profiles were also altered,indicating cell cycle arrest and inhibition of cell proliferation.Administration of DZNep in immunodeficient mouse xenograft models led to significant reduction of tumour volume, especially at higher doses.Subsequent immunohistochemical analyses revealed inhibition of cell proliferation and induction of apoptosisin vivo[62].
In leiomyosarcoma cell lines, treatment with EPZ011989 resulted in a noticeable decrease in H3K27me3 levels compared to control.Furthermore, in BEZ235-resistant cells prior treatment with EPZ011989 significantly increased their sensitivity to BEZ235 (dactolisib, a dual PI3K/mTOR inhibitor) and a reduction of cancer stem cells (CSCs).In xenografts, pre-treatment with EPZ001989 in combination with BEZ235 led to a significant reduction in tumour growth compared to either drug alone[63].
Treatment of well-differentiated and dedifferentiated liposarcoma cell lines with GSK343 led to reduction of cell proliferation, followed by a decrease in levels of H3K27me3[41].Similarly, combination therapy using GSK126 and 5-aza-dC, a chemical analog of cytidine, in a dedifferentiated cell line showed anti-proliferative effects with a subsequent increased apoptosis and enhanced expression of adipocytes-specific differentiation genes[64].These findings suggest combining epigenetic drugs may be more effective than single agents alone.
A number of clinical trials involving STS patients treated with small molecule inhibitors of EZH2 are currently ongoing.These include treatment with tazemetostat of patients with SS, ES or SMARCB1-deficient tumours (NCT02601937, NCT02875548 and NCT02601950).The combined approach of tazemetostat and doxorubicin is also being tested in a clinical trial for patients with advanced ES(NCT04204941).
THE MECHANISM OF ACTION OF EZH2 IN STS
It must be emphasized that EZH2 is a ubiquitous and multifaceted enzyme, involved in numerous regulatory axis and molecular networks, including those promoting oncogenesis.Vella and colleagues demonstrated in ERMS that the levels of a number of miRNAs were restored as EZH2 levels decreased,resulting in anti-cancer activity.Furthermore, depletion or inhibition of EZH2 was highly correlated with altered cell cycle and reduction of expression of several proteins, such as Ki-67 or TBX3 in RMS.However,the role of EZH2 should be considered differently depending on the cellular context: it can serve as a transcriptional suppressor and a transcriptional co-activator[65,66].Previous research has shown that EZH2 may act in noncanonical manner, independently of PRC2 and its histone methyltransferase function,activating the downstream genes through non-histone targets methylation or direct binding to proteins in several cancer types[67-70].Additionally, the PRC2-SWI/SNF interactions are essential in the regulation of gene expression in several signalling pathways, such as RB, Cyclin D1, MYC and hedgehog, which are impaired in many cancer types[6].In the presented results, EZH2 depletion led to decreased activity of the oncogenic molecules such as E2F and c-Myc involved in signal transduction pathways[39], proposing indirect implications of EZH2 to oncogenesis.When interpreting the outcomes, there is also a need for contextualizing these with relation to the tumour microenvironment (TME), which consists not only of cancer cells but also of extracellular matrix, fibroblasts, adipocytes, endothelia, and immunomodulators such as T and NK cells, tumour-associated macrophages and dendritic cells.All of these play an essential role in the overall response to the therapy and contribute to tumour resistance[71].Furthermore, the diversity and number of reactions within the TME may promote cancer escape from immune vigilance[72].Having said this, the complexity of TME may be one of the main reasons for the diverse results among different STS and even within the same histology.Another likely explanation may be heterogenous character of STS,various kinetics and attributes of the cell lines or xenografts, specificity of the inhibitor used and underlying genetic alterations.
The role of PRC2/EZH2 in the immune response - a potential combination therapy?
The involvement of PRC2/EZH2 in the immune response is a burgeoning area of research that has implications for both immune-based therapy efficacy and use of EZH2 inhibition as a treatment itself.Immune therapy is not yet a common modality in STS; however, combination therapies utilising EZH2 inhibition and immune therapies may prove to be an effective treatment for STS.
PRC2/EZH2 has been shown to be involved in normal haematopoiesis, the regulation of immune cells, and is essential for T-cell proliferation and anti-tumour immunity[73-76].Conversely it is also implicated in some haematological malignancies[77,78].One potential complication of using EZH2 inhibitors as single agents is unintended effects upon the immune environment.A paper by Huang and colleagues in 2019 identified that in immune-competent mice treated with GSK126, the anti-tumour effects of EZH2 inhibition were reduced compared to immune-deficient mice[79].Importantly the authors also described increased production of myeloid derived suppressor cells (MDSCs) in the GSK126 treated, immune-competent mice, potentially leading to an immune suppressive microenvironment.The authors suggest this as a mechanism behind some of the poorer trial results seen with GSK126.
On the other hand, there is evidence that inhibition of EZH2 may help to improve anti-tumour responses to specific immune check point inhibitors.For example, CPI1205 has been shown to improve the efficacy of the anti-CTLA4 treatment, ipilimumab in a mouse model, primarily by compensating for an increased level of EZH2 induced by ipilimumab itself.The addition of CPI1205 was found to modulate cytotoxic T cells and phenotypically alter Tregs into effector-like T cells and improve the immune response[80].Furthermore,Xiaoet al.(2019)[81]showed thatin vitroEZH2 negatively regulated the expression of both PD-L1 (CD274)and the transcription factor, interferon regulatory factor 1 in hepatocellular carcinoma (HCC) cells and clinical samples.The authors posited that an EZH2 inhibitor may therefore improve responses to anti-PD-1/PD-L1 therapy.
In short, EZH2 as a treatment target will certainly be of use in immune therapies; however, due to its role in both normal and abnormal immune processes it is likely that its efficacy will be sub-type/genotype dependant, highlighting the need for studies into this potential combination for the treatment of STS.
CONCLUSIONS
This review has evaluated EZH2 as a potential target in treatment of STS, an exceptionally diverse group of malignancies, with various underlying genetic and epigenetic alterations defining their development, clinical behaviour and responsiveness to therapies.However, one limitation of this review is that many STS have yet to be studied in the context of EZH2.There are over 60 distinct histological subtypes of STS, for many of which we have not yet understood the contribution of EZH2 or how it may be exploited for therapeutic benefit.Table 2 summarises the main findings of this review.
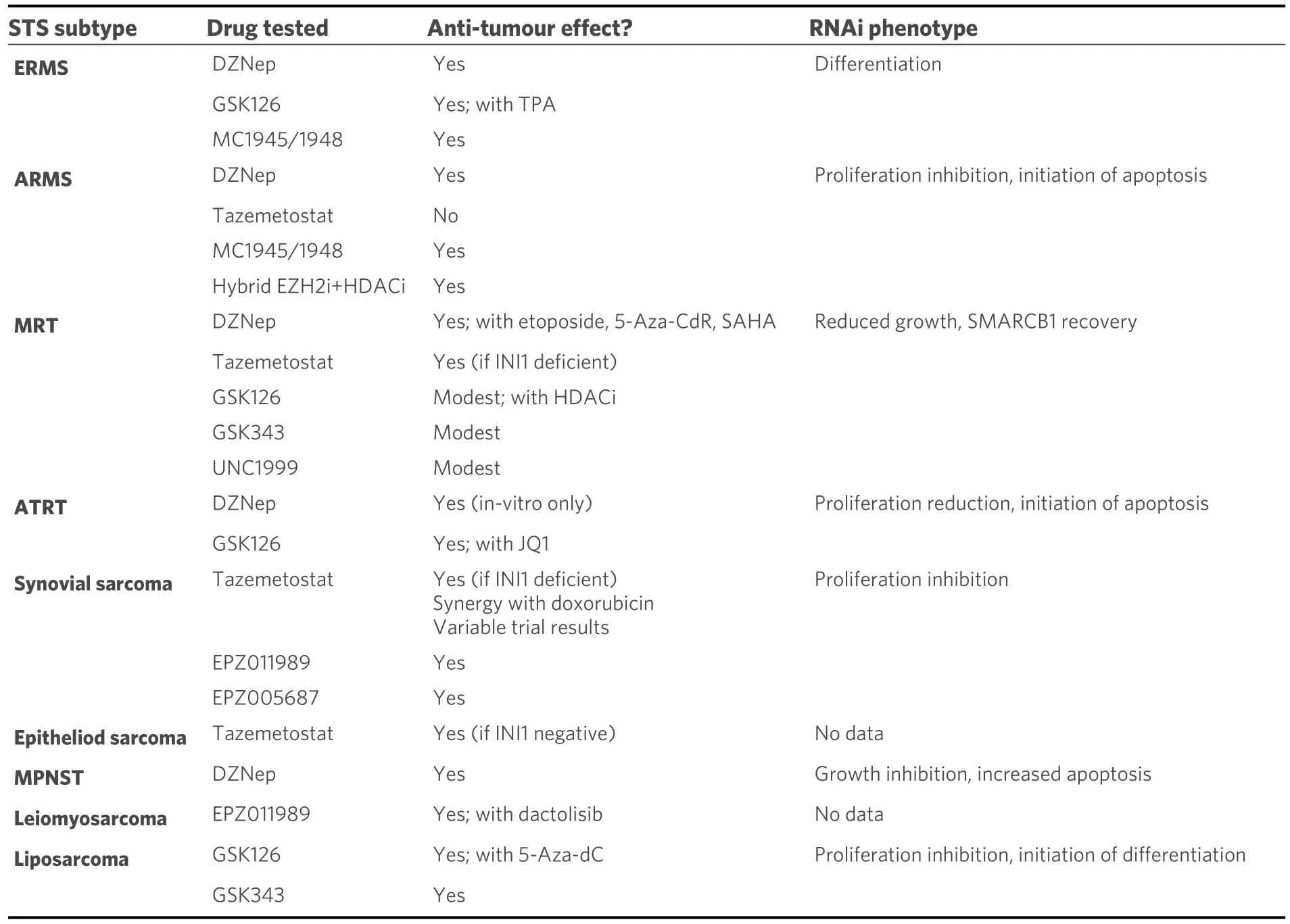
Table 2.A summary table of the STS highlighted in this review and the effects of EZH2 modulation for each
STS reviewed in this study almost ubiquitously express high levels of EZH2.As there is no evidence of genetic aberration of EZH2 that might be contributing to this overexpression, it is highly likely that this is the result of the undifferentiated nature of STS, whereby STS cells retain hallmarks of their likely cell of origin, a mesenchymal stem cell.For certain STS, the genetic hallmark of SMI/SNF deregulation,exemplified by SMARCB1 deficiency seen in SS, is likely contributing to the aberrant activity of PRC2 driven by EZH2, which in turn is thought to predispose tumour cells to sensitivity to EZH2 inhibition[52].Indeed, in SS the majority of the cases exhibit lack of SMARCB1 protein as well as the presence of SS18-SSX translocation, which is thought to make cells more prone to EZH2 inhibition.
The correlation of EZH2 protein expression with clinical features of STS is not consistent across studies.However, the most common trend seen includes highly enhanced levels of protein in malignant tumour/cell lines and metastatic lesions, as compared to benign tumour or healthy tissue.The highest divergence arose among EZH2 overexpression with regard to histological grade and clinical stage of the tumours, suggesting either limited sample size or patient/tumour specific EZH2 expression in each phase of the disease.
This study has highlighted both similarities and differences in EZH2 expression and response to EZH2 modulation across STS subtypes.Each STS type has a differing cell lineage type, leading to differing genetic and epigenetic diversity both between STS types and even within STS subtypes.As a consequence, this may explain differences in response to EZH2 modulation as subset of genes that are both targeting and are targeted by PRC2/EZH2 are likely to be typical to that cell type.This may therefore also have implications in the use of EZH2 inhibitors as therapeutics and thus differing combinations with EZH2 inhibitors need to be investigated for different STS.
The results of pharmacological and genetic inhibition of EZH2 seem to be highly promising as a therapeutic option for STS.One key aspect is inhibitor specificity.DZNep is the least specific for EZH2 as its mode ofaction is inhibition of S-adenosylhomocysteine hydrolase and therefore also inhibits the activity of other methyltransferases[82].Conversely, tazemetostat has been shown to be highly potent and selective for EZH2[52].Worth noting is the fact that combined treatment of specific EZH2 inhibitors either with differentiating agents, other epigenetic agents or chemotherapeutics resulted in enhanced anti-tumour activity in many STS studies.A number of STS are predominantly associated with paediatric onset and thus strategic combination of EZH2 targeted inhibitors with standard of care therapies will likely be the most effective course of treatment for many STS.
DECLARATIONS
Acknowledgments
Not applicable.
Authors’ contributions
Made substantial contributions to conception and design of this review: Karolak M, Walters ZS
Original draft preparation: Karolak M, Walters ZS
Writing, review, and editing of manuscript: Karolak M, Tracy I, Shipley J, Walters ZS
Performed literature research: Karolak M
Preparation of figures: Tracy I, Walters ZS
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by a Children with Cancer UK grant (Walters Z S) and by Sarcoma UK (Shipley J,Walters ZS)
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2021.
 Journal of Cancer Metastasis and Treatment2021年3期
Journal of Cancer Metastasis and Treatment2021年3期
- Journal of Cancer Metastasis and Treatment的其它文章
- Diverse roles of bitter melon (Momordica charantia)in prevention of oral cancer
- Leelamine suppresses cMyc expression in prostate cancer cells in vitro and inhibits prostatecarcinogenesis in vivo
- Seaweeds’ nutraceutical and biomedical potential in cancer therapy: a concise review
- Role of 18F-FDG PET/CT in detection of hematogenous metastases of advanced differentiated thyroid carcinoma: a systematic review and meta-analysis