
Autophagy and thyroid cancer
2021-07-19 12:30YujiNagayama
Yuji Nagayama
Department of Molecular Medicine,Atomic Bomb Disease Institute,Nagasaki University,Nagasaki 852-8523 Japan.
Abstract This review provides the up-to-date physiological,pathophysiological,and carcinogenic roles of autophagy in the thyroid.The data on its physiological roles are mainly obtained with genetically engineered mice,demonstrating the importance of autophagy for the maintenance of cell homeostasis and survival as well as provision of building blocks for sufficient synthesis of proteins such as thyroglobulin.Positive and negative controls of autophagic activity by thyrotropin and thyroid hormone,respectively,are now apparent.In thyroid cancer,there is no published study on the role for autophagy in the initiation/development of thyroid cancer,and there exist many inconsistent data regarding its role in established thyroid cancer behavior.For example,definitive conclusions remain to be elucidated regarding the level of autophagic activity in thyroid cancer cells and the effect of autophagy inducers/inhibitors on cancer cell survival/proliferation.Especially,when autophagy is targeted in novel cancer therapeutics,some studies show its pro-survival,but others its anti-survival or context-dependent,effects on thyroid cancer cells.Further studies are in the future necessary to further elucidate the potential of autophagy as a therapeutic target for thyroid cancer.
Keywords: Thyroid,cancer,autophagy,pro-survival,anti-survival
INTRODUCTION
Thyroid cancer is the most common endocrine malignancy with its incidence rate being rapidly increasing during the last several decades[1].The extensive studies on genetic/genomic landscapes of thyroid cancers have identified the aberrant activation of the RAS-RAF-MEK-ERK signaling pathway by the point mutations in BRAF or RAS,or chromosomal rearrangements like RET/PTC as the causative role in thyroid carcinogenesis[2].Thyroid cancer can be divided into two subgroups -differentiated [papillary (PTC) and follicular (FTC)] and de-differentiated [poorly differentiated and anaplastic (ATC)] types.PTC is the most common type,in which the mutant BRAF (BRAFV600E) has been shown to be the most frequent driver mutation,followed by RET/PTC and the mutant RAS[2].Surgery,radioactive iodine (RAI) treatment with131I and thyrotropin (TSH) suppression therapy have long been choices of treatment modalities for thyroid cancer.In most patients with thyroid cancers,disease remission is achieved by these modalities,but recurrence does occur in a subgroup of patients who acquired RAI resistance as a result of dedifferentiation.Tyrosine kinase inhibitors have recently been approved as an additional therapeutic choice for thyroid cancer treatment,especially for RAI refractory thyroid cancer,which clearly exhibit its therapeutic benefits,but at the same time show some problems such as severe adverse effects and/or intrinsic/acquired resistance.Thus,more novel therapeutic approach is urgently needed to be introduced.Targeting autophagy may have a potential for this purpose.
Autophagy is an essential pathway mediating the degradation of cellular components such as proteins and organelles in the lysosomes.It not only occurs constitutively at basal rate under physiological conditions to maintain homeostasis for intracellular recycling of the metabolites and metabolic regulation,but it is also induced during various physiological and pathological conditions such as nutritional starvation[3,4].Alterations in autophagic activity have been widely considered to play crucial roles in numerous diseases such as degenerative disorders,metabolic diseases,aging,and cancer,among others.There are three types of autophagy so far identified in mammalian cells:macroautophagy (hereafter referred to as “autophagy”),microautophagy,and chaperone-mediated autophagy (CMA).This review mainly focuses on macroautophagy.
AUTOPHAGY IN PHYSIOLOGY
Autophagic flux
In the autophagy process,a special structure called the “phagophore” is first formed from the endoplasmic reticulum or other intracellular membranes in a cell,which engulf the cellular contents and targeted proteins/organelles.Upon the complete engulfment by its enclosure,it becomes the “autophagosome” and fuses with the lysosome forming the “autolysosome” and then delivers its contents into the lumen of the lysosome for degradation.There are two kinds of autophagy,non-selective,and selective.Nonselective autophagy engulfs a part of the cytoplasm as cargo,while selective autophagy recognizes a specific cargo such as damaged organelles/protein aggregates;an example is mitophagy -selective autophagy for mitochondria.The highly simplified schema depicting the autophagic flow and a representative autophagy inducer [rapamycin,an inhibitor for mechanistic target of rapamycin (mTOR)] and inhibitors [3-methyladenine (3-MA) and chloroquine (CQ)] is shown in Figure 1.Although more than 30 proteins are involved in autophagy,most are omitted and only those appear in this review are shown in Figure 1.For more details,refer to the following references[3,4].
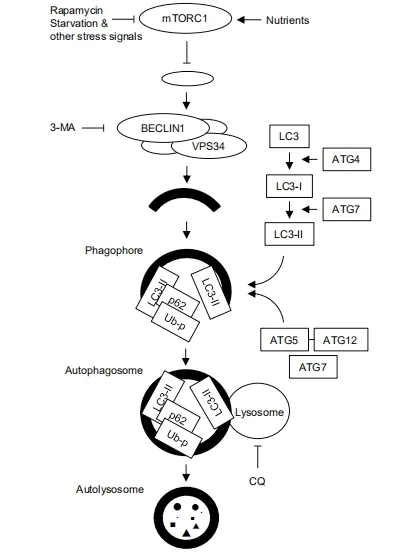
Figure 1.Schematic representation of autophagic flux.Autophagy starts with formation of phagophore (the initiation step),followed by the nucleation step where class III phosphatidylinositol 3-kinase (PI3K) complex,containing BECLIN-1,VPS34,and other molecules,elongates phagophore and encapsulates the cargo that should be degraded,result in formation of autophagosome.The next elongation step involves two ubiquitin like conjugation systems -the LC3-PE and ATG5-ATG12 systems.In the former,processed and lipidated LC3 (LC3-II) is recruited to the phagophore/autophagosome,as is p62,an adaptor molecule for selective autophagy of ubiquitinated proteins (Ub-p).The latter is essential for proper curvature of the phagosome.ATG7 is involved in both systems.The autophagosome then fuses with the lysosome to form autolysosome,in which the cargo is degraded by the lysosomal enzymes.mTOR negatively controls autophagic activity,whose activity is controlled positively by nutrients and negatively by rapamycin and other stress signals such as starvation.3-MA inhibits autophagy at the early step of autophagic flux by inhibiting class III PI3K,and chloroquine at its late step by raising the lysosomal pH and then inhibiting the fusion between the autophagosome and the lysosome.LC3:light chain 3;CQ:chloroquine;3-MA:3-methyladenine
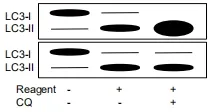
Figure 2.Alterations in relative amounts of LC3-I and II by autophagy inducer and inhibitor in Western blotting.A reagent increases amounts of LC3-II in both upper and lower panels.However,a further increase in LC3-II by combination of the reagent and chloroquine indicate that the reagent is an autophagy inducer,but when the reagent is an inhibitor,amounts of LC3-II remains unchanged in combination of the reagent and chloroquine.LC3:light chain 3;CQ:chloroquine
Monitoring of autophagic flux
It is important to precisely measure autophagic activity to correctly understand the role for autophagy in pathophysiology.This is very critical because the appropriate analyses have not always been done in many previous studies on the role for autophagy in thyroid cancer (see “NOTES” below).
To correctly evaluate autophagic activity,flux through autophagy pathway should be measured.Mere determination of the amounts of autophagy-related proteins does not necessarily suffice this purpose.Microtubule-associated protein 1 light chain 3 (LC3) and p62/sequestosome 1 (SQSTM1,an adaptor molecule for selective autophagy) are two representative molecules among those used as biomarkers for monitoring autophagic flux[5].As autophagy proceeds,LC3-I,ubiquitously expressed in the cytoplasm,is converted to LC3-II by lipidation,conjugated with phosphatidylethanolamine (PE) and possibly phosphatidylserine,and is recruited to the autophagosome.The amount of LC3-II can be monitored by Western blotting (WB) showing the presence of a LC3-II band in addition to a LC3-I band,and by immunohistochemistry or immunofluorescence showing alteration of LC3 staining pattern from diffuse to punctate appearance.Although the molecular weight of LC3-II is higher than that of LC3-I (16 to 18 kDa) because of addition of lipid,LC3-II migrates faster in gel due to its high hydrophobicity and can be detected as a 14 to 16 kDa band in WB.However,at the same time,LC3-II is degraded by the lysosomal digestion in autophagic flux,so that the amount of LC3-II is determined by balance between the rate of conversion from LC3-I to LC3-II and degradation in the lysosome.p62 is a typical substrate of autophagy and is degraded in the autolysosome.The amounts of p62 can also be determined by WB and immunohistochemistry or immunofluorescence.
Thus,in a typical case,increased amount of LC3-II and decreased amount of p62 indicate elevated autophagic flux.In the case of autophagy inhibition,the alterations in the amounts of LC3-II and p62 depend on which step is inhibited.For example,3-MA,which blocks autophagy at its initial step[6],decreases LC3-II and increases p62,but CQ,which blocks autophagy at its late step[7],increases both LC3-II and p62.However,what is complicated is that the reverse of the latter is not always true,that is,increased LC3-II and p62 does not necessarily mean autophagy inhibition.This is because the p62 level is controlled not only by its degradation by autophagy but also by its synthesis rate;when synthesis rate exceeds degradation rate,p62 levels may increase even if autophagy is active.Indeed,transcriptional/posttranscriptional increases in p62 levels are reported in certain conditions[8-11].Therefore,an additional experiment in which the cells are treated with a reagent of your interest,an autophagic inhibitor (which inhibits autophagy at the late step like CQ) and their combination for comparison of LC3-II levels should be performed to confirm its effect on autophagic flux (see references[5,12]for more details).Briefly,an additive or supra-additive effect of the combination on LC3-II levels likely indicate enhanced effect of autophagic flux by the reagent,while no additive effect suggests a blockade of autophagy [Figure 2].
The plasmids expressing green fluorescence protein (GFP)-LC3 or red fluorescence protein (RFP)-GFPLC3 are also frequently used for autophagy flux monitoring[5].GFP puncta in immunofluorescence represent autophagosome,not autolysosome,formation with the GFP-LC3 vector because GFP fluorescence is quenched in low pH of the lysosome,which therefore indicate formation of autophagosome and does not indicate completion of autophagy flux.In contrast,in the RFP-GFP-LC3 vector,yellow color,signifying co-localization of red and green fluorescence,indicates formation of autophagosome while red indicates formation of autolysosome where red is preserved but green is quenched in the lysosome;the latter indicates completion of autophagic flux.
Physiological roles for autophagy
In general
Indispensable role for basal level of autophagy for maintaining homeostasis was clearly shown systematically by genetically engineered mice.Thus,systemic disruption ofAtg5orAtg7genes caused neonatal death,that is,the knockout (KO) mice died within a day after birth[13,14].Organ-specific KO induced tissue degeneration in neuron,liver,heart and pancreatic β-cells[15-18].The crucial role for autophagy in development and differentiation of numerous other tissues have also been reported[3].These data clearly indicate the principal role for autophagy in development and differentiation of many different types of cells.However,even in the physiological condition,it is also true that excessive or prolonged autophagy leads to cell death called “autophagy-regulated cell death”[19,20].
In thyroid
The physiological role for autophagy in thyroid was studied using genetically engineered mice.We generated the thyroid specificAtg5,a component of the autophagy machinery [Figure 1],KO miceAtg5flox/flox;TPO-Cre(Atg5thyr-KO)[21].These KO mice were born with an expected Mendelian trait and in euthyroid state during the 1-year-observational period.However,accumulation of ubiquitinated proteins was detected at 4 months,and apoptotic cell death,decreased number of thyroid follicular epithelial cells (thyrocytes),and increased DNA damages detected by 8-hydroxy-2’-deoxyguanosine (8-OHdG) and p53-binding protein 1 (53BP1) foci were observed at 8 and 12 months,indicating the critical role of basal level of autophagic activity in thyrocyte survival and homeostasis.
To study the significance of induced autophagy in a stressed condition,we first clarified regulation of autophagic activity by hormones,finding the positive control by TSH and the negative control by thyroid hormone T4[11].When KO mice were treated with methimazole and perchlorate,which induce increased TSH and decreased T4levels (as a net effect,induce autophagy),smaller follicle sizes and lower thyroglobulin (TG) contents in thyrocytes were observed as compared to control mice,implying impaired TG production presumably by diminished nutrient supply due to a lack of autophagy.Altogether,basal level of autophagy is required for maintenance of cell homeostasis and survival,and induced autophagy,elicited by elevated TSH and decreased thyroid hormone,supports the provision of building blocks for sufficient synthesis of proteins including TG.
KO mice with disruptedVps34gene,a component of class III PI3K (a more upstream molecule than ATG5 in autophagic flow;see Figure 1) were also generated[22].Vps34flox/flox[11];Pax8-Cre(Vps34cKO) mice were also born normally but died at approximately 1 month of age;they had severe hypothyroidism,impaired TG iodination,abnormal TG trafficking,defective lysosomal proteolysis,and the presence of macrophages eating iodinated TG in the lumen were observed.More pronounced phenotype alterations inVps34cKOmice are evident when comparing toAtg5thyr-KOmice,which may be explained by autophagy-independent functions of autophagy-related proteins[23].Indeed,the findings in the latter seem to be at least in part attributed to the role for VPS34 in vesicular trafficking rather than autophagic activity.
AUTOPHAGY IN CANCER
Cancer initiation
In general
It is well accepted that autophagy has the dual effects on cell survival and death,that is,it on one hand supports cell survival by providing nutrients required for maintaining cellular homeostasis,but on the other hand promotes cell death,i.e.,autophagy-regulated cell death[19,20].Particularly in cancer,it is proposed that autophagy suppresses tumor development in the early stage of tumorigenesis,while autophagy promotes tumor survival and proliferation once a tumor develops[24].Therefore,numerous preclinical and clinical studies on autophagy-targeted treatment mainly focused on autophagy inhibitors[24,25].However,the data so far accumulated are not so simple,and even in established cancers,studies show therapeutic effect of not only autophagy inhibitors but also autophagy inducers on tumor growth[24,26].
Much evidence indicating the role for autophagic activity in the initial step of cancer development comes from the studies with genetically engineered mice.Beclin-1heterozygous KO mice developed spontaneous lung cancers,liver cancers,and lymphomas,and accelerated the development of HBV-induced liver premalignant lesions[27,28].Systemic mosaicAtg5KO and liver-specificAtg7KO mice developed benign liver adenomas[29,30].Although accumulated p62,commonly observed in all KO mice,is thought to play a role for tumorigenesis[30,31],development of cancers in multiple organs inBeclin-1heterozygous KO mice as compared to that of benign tumors only in liver in systemic mosaicAtg5KO and liver-specificAtg7KO mice may indicate autophagy-independent anti-tumorigenic function of BECLIN-1;for example,BECLIN-1-controls of apoptosis[32]and of p53[33].It is also reported thatAtg4KO itself did not induce any tumor development,but increased incidence of chemically induced fibrosarcoma[34].
Of interest,Atg5orAtg7KO mice were crossed with other genetically engineered mice having various oncogenes or a loss-of-function mutation of tumor suppressor gene such as the mutant RAS,BRAF or p53[35].In most of these double mutant mice,tumor development was accelerated but tumors did not progress to malignancy and,instead,turned to be benign oncocytic tumors[36-40].Oncocytic tumor has abundant mitochondria,which is thus likely attributed to defective mitophagy,a selective autophagy.Therefore,impaired mitophagy and/or accumulated mitochondria appear to block progression from benign to malignancy.Accumulation of deformed and dysfunctional mitochondria was also observed in liver adenomas in liver-specificAtg7KO mice mentioned above[29,30].
Altogether,it is clear that autophagy works as a tumor suppressor in the early step of tumorigenesis.However,there is always an exception;development of some tumors such as RAS-driven cancers are reported to be dependent on autophagy activation[41].
Impaired autophagy is also strongly associated with abnormal DNA damage repair[42],especially abnormal homologous recombination[43],which may also explain the propensity of autophagy-defective mice to tumor development.Indeed,genomic instability was suggested by the presence of γH2AX foci (indicating DNA double strand breaks) in liver adenomas inAtg7KO mice[29,30].
In thyroid
In thyroid cancer,there is no published study on cancer development using autophagy KO mice.In our preliminary study,no thyroid tumor development was observed in thyroid-specific autophagy KO mice (Atg5thyr-KO) at 12 months nor inAtg5flox/floxmice subjected to intrathyroidal injection of adenovirus expressing Cre recombinase under TG promoter in the 18-month-experimental period (unpublished data).Instead,a study on a human genetic disease provides a clue as to the relationship between autophagy and thyroid carcinogenesis.Thus,a previous study has shown that,in human Cowden syndrome with the mutant succinate dehydrogenase D-G12S or D-H50R,suppression of autophagy by these mutations and subsequent ROS elevation and phosphatase and tensin homolog (PTEN) inactivation may be involved in thyroid tumorigenesis[44].Furthermore,SNPs in autophagy-related genes such asAtg5andAtg16Lare reported to be associated with development of thyroid cancer and RAI resistance[45,46].
As mentioned above,oncocytoma is usually a benign tumor.However,the thyroid is an exception,where it is sometimes malignant (called Hürthle cell carcinoma).In this regard,we have recently found that accumulation of mitochondria in thyroid oncocytic tumor is caused (1) by defective expression of mitochondria-eating protein (MIEAP)[47],which play a central role in a non-canonical mitophagy[48];and (2) by enhanced biogenesis of mitochondria which compensates defective function of mitochondria complex I[49].It is reported that the defective PARK2-mediated canonical mitophagy by a loss-of-function mutation inPARK2gene also causes mitochondrial accumulation in a thyroid malignant oncocytoma cell line XTC.UC1,but loss of function mutation in autophagy-related genes includingPARK2is rare in thyroid oncocytoma in humans[50,51].Therefore,in experimental mouse models of cancer in nonthyroid tissues (see above),a defect in canonical mitophagy may be sufficient,but in human thyroid,both defective mitophagy (i.e.,a defect in either MIEAP-mediated non-canonical or PARK2-mediated canonical mitophagy) and increased compensated mitochondrial biogenesis may be required for oncocytic phenotype.
Established cancer
Autophagic activity in thyroid cancer
In established cancers,irrespective of those derived from the thyroid or other organs,it is controversial whether autophagic activity is elevated or not.In thyroid cancers,autophagic activity is reported lower in PTCs,especially those with lymph node metastasis,by demonstrating lower LC3-II expression levels in WB (NOTE -p62 levels were not studied.)[52]and by lower expression levels of LC-3 protein in immunofluorescence (NOTE -LC3 puncta,not LC3 expression levels,should be quantified in immunofluorescence)[53],while no difference was found in number of LC3 puncta in immunofluorescence between benign (including normal,Graves’,goiter,and follicular adenoma tissues) and malignant (including FTC,PTC,follicular variant PTC,and ATC) lesions[54].Lower autophagic activity in thyroid cancer can be explained by the fact that the oncogene-stimulated RAS-RAF-MEK-ERK and mTOR signaling pathways inhibit autophagy[55,56].In general,oncogenic proteins inhibit,but oncosuppressor proteins induce autophagy[57].On the other hand,BRAF-activated long non-coding RNA (BANCR),which is overexpressed in PTCs,contributes to cell proliferation and activates autophagy as demonstrated by increased and decreased LC3-II levels by overexpression and knockdown (KD),respectively,of BANCR in WB (NOTE -p62 levels were not studied)[58].BRAFV600E-induced chronic endoplasmic reticulum stress[59]and NOXA upregulation are also thought to be reasons for BRAFV600E-mediated autophagy activation in melanoma[60].The associations of BRAFV600Eand higher expression levels of autophagy-related proteins (LC3 and p62)[61]and higher expression of BECLIN-1[62]are noted in PTCs.Up-regulation of LC3-II expression is also reported to be associated with hypoxia-inducible factor-1α (HIF-1α) and telomerase reverse transcriptase (TERT) in PTCs[63].Of note,higher expression levels of autophagy-related proteins do not necessarily mean higher autophagic activity.
Finally,CMA is shown increased in PTCs as demonstrated by overexpression of lysosome-associated membrane protein type 2A (LAMP2A) (NOTE -the flux of this type of autophagy cannot be examined)[64].
Autophagy as biological markers for thyroid cancer
Different groups report the distinct autophagy-related gene sets as biological markers for thyroid cancer.Zhuet al.[65]reported three genes related to prognosis:inositol 1,4,5-triphosphate receptor type 1(ITPR1),chemokine C-C motif ligand 2(CCL2) andcyclin-dependent kinase inhibitor 2A(CDKN2A).Zhanget al.[66]reported two genes related to aggressiveness and prognosis:BECLIN-1andaplasia Ras homolog member I(ARH1).A positive correlation between LC3-II levels and urinary iodine concentrations is also reported in thyroid cancer patients[67].
Autophagy-targeted therapy
Regarding the results of autophagy-targeted therapy,reports can be divided into three categories;the first supporting pro-survival/proliferative effect of autophagy on thyroid cancer cells [Table 1],second supporting anti-survival/proliferative effects [Table 2],and the third suggesting cell-context-dependent effects [Table 3].
(1) Studies reporting pro-survival/proliferative effect of autophagy on thyroid cancer
In the first group demonstrating the pro-survival effect of autophagy on thyroid cancer cells [Table 1],anti-tumor effects of autophagy inhibitors -CQ,3-MA,and genetic KD of autophagy related proteins -are inconsistent;CQ is effective in 8505C,FTC133,and TPC1 cells[63,68]as well as BCPAP cells[69](concentration used:10 μM),but not in BCPAP and FRO cells[70],KHM-5M and C643 cells[71],and K-1 cells[72](10-20 μM).Another autophagy inhibitor 3-MA is reported to suppress cell growth in BCPAP cells[67](5 mM),but not in KHM-5M and C643 cells[71],TPC1 cells[73],SW579 cells[74],and BCPAP[75,76](1-3 mM).Genetic ablation of autophagy byATG5KD was also shown to kill 8505C and FTC-133 cells[68],and byATG7KD in BCPAP and TPC1 cells[63,69].It should be noted here that CQ has also been reported to have an autophagyindependent anti-cancer effect[77-79].
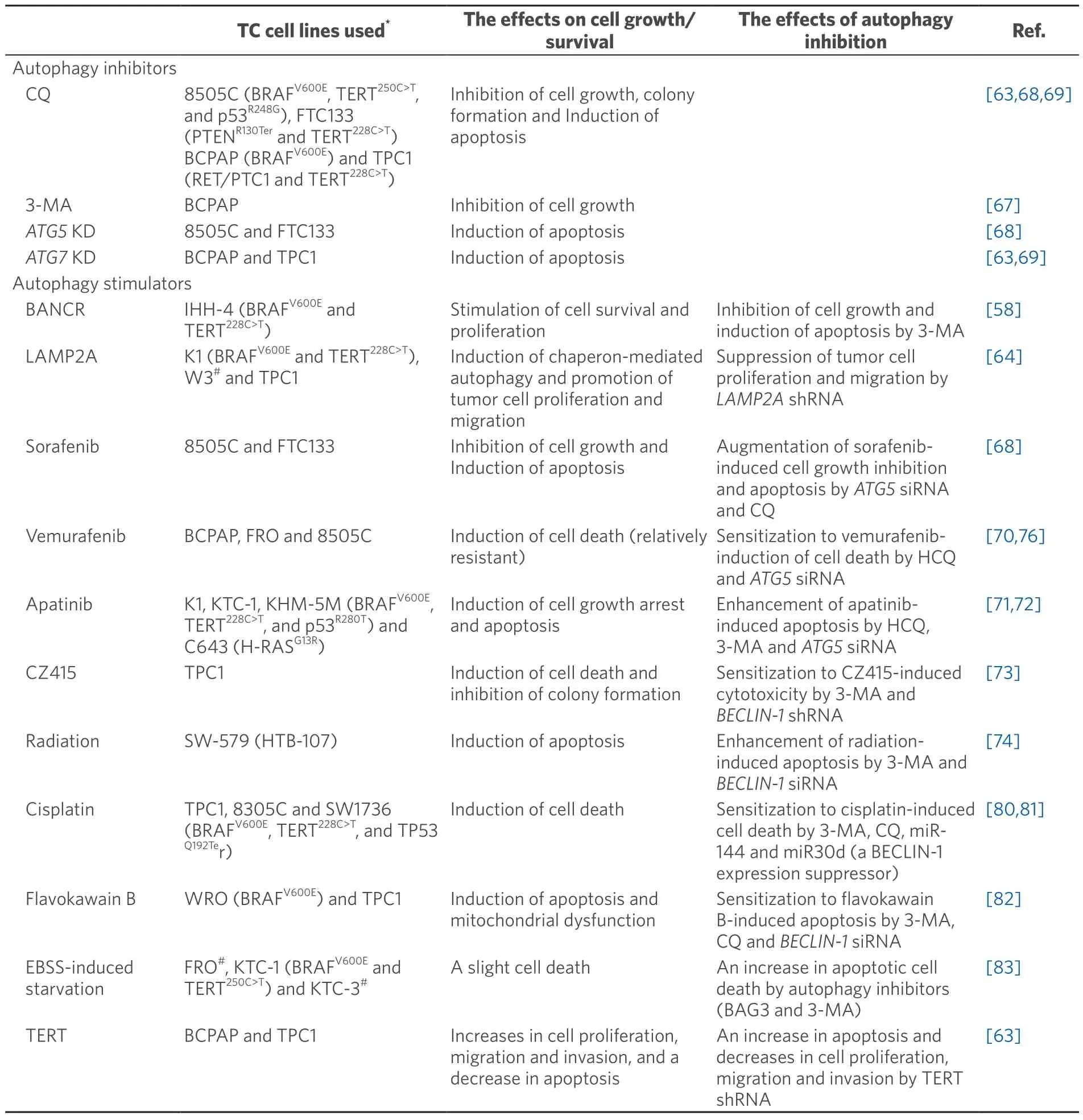
Table 1.Summary of the articles reporting pro-proliferative/tumorigenic action of autophagy
Autophagy inducers so far reported to show the pro-survival/proliferative effect of autophagy include BANCR[58],LAMP2A[64],tyrosine kinase inhibitors [sorafenib (a pan-tyrosine kinase inhibitor) and vemurafenib (a BRAF inhibitor)])[68,70,76],apatinib (a VEGF2 inhibitor)[71,72],CZ415 (an mTOR inhibitor)[73],radiation[74],cisplatin[80,81],flavokawain B[82]Earle’s Balanced Salt Solution-induced starvation[83],and TERT[63][Table 1].NOTE -among them,some data are not convincing.For example:(1) although the authors interpret their data showing augmentation of sorafenib-induced apoptosis by ATG5 KD in thyroid cancer cell lines 8505C and FTC133 as indicating the pro-survival effect of autophagy,sorafenib and ATG5 KD individually induced apoptosis and the effect of their combination look merely additive (or less than additive)[68];(2) autophagic activity was not evaluated in the CZ415 study[73];and (3) only LC3-IIlevels were determined in WB in the BANCR study[58].Tyrosine kinase inhibitors are recently approved as a new thyroid cancer therapeutic regimen,especially RAI-refractory thyroid cancer.In this regard,the relationship between tyrosine kinase inhibitors’ anti-cancer effect and autophagy is clinically of interest,but the data on vemurafenib (CQ or 3-MA alone showed no cytotoxic effect but enhanced the vemurafenib’s effect[70,76]) and sorafenib (see the NOTE above[68]) are not consistent.More complicatedly,there is a report that vemurafenib-induced autophagy is anti-survival in melanoma cells with BRAFV600E[84]that is otherwise pro-survival[83]in thyroid cancer cells as mentioned above[70].
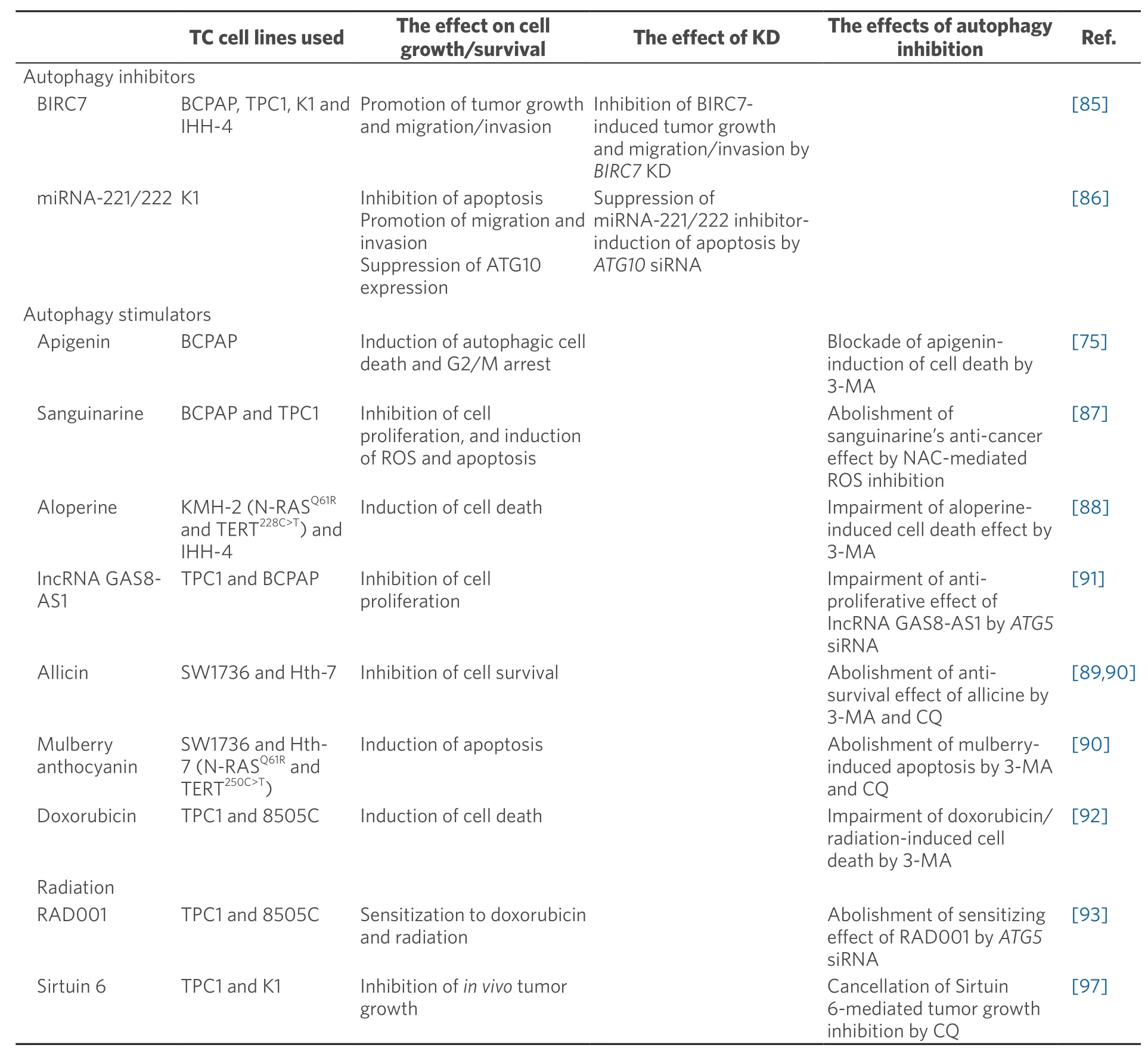
Table 2.Summary of the articles reporting anti-tumorigenic action of autophagy

Table 3.Summary of the articles reporting context-dependent effect of autophagy modulators on cancer behavior
(2) Studies reporting anti-survival/proliferative effect of autophagy on thyroid cancer
On the other hand,there are also some articles reporting the anti-survival/proliferative effects of autophagy on thyroid cancers [Table 2],which include autophagy inhibitors of baculoviral IAP repeat containing 7 (BIRC7)[85]and miRNA-221/222[86],and autophagy inducers such as natural compounds (e.g.,sanguinarine[87],aloperine[88],allicin[89],mulberry anthocyanin[90],and apigenin[75]),long non-coding RNA GAS8 antisense RNA1 (lncRNA GAS8-AS1)[91],doxorubicin[92],radiation[92],RAD001 (everolimus,an mTOR inhibitor)[93],and Sirtuin 6[94].NOTE -(1) although Linet al.[92]used doxorubicin as an autophagy inducer because it increased LC3-II levels (p62 levels were not studied),it is shown in other papers that doxorubicin blocks autophagy at the late stage (therefore increases both LC3-II and p62,like CQ) by impairing lysosomal acidification[95-97];(2) the interpretation of the data on aloperine is difficult -alopeirne increased both LC3-II and p62 levels but further increase in LC3-II could not be seen in combination of aloperine and CQ indicating aloperine-inhibition of autophagy as mentioned earlier,while the red fluorescence in aloperine-treated cells and yellow in aloperine and CQ-treated cells transfected with the mRFP-EGFP-LC3 vector suggest aloperine-induction of autophagy[88];and (3) the effect of anthocyanin on autophagy was only studied by LC3-II levels in WB and formation of autophagosome by electron microscopy and the GFP-LC3 vector but not by p62 levels or formation of autolysosome[90].
Inconsistent data between Tables 1 and 2 include (1) mTOR inhibitors CZ415 and RAD001 show the opposite effects,pro-survival using TPC1 in the former[73]and anti-survival using TPC1 and 8505C cells in the latter[93];and (2) radiation is also reported to be pro-survival in SW-579 cells[74]and anti-survival in TPC1 and 8505C cells[92].
(3) Studies reporting context-dependent effect of autophagy on thyroid cancer
The third group,context-dependent effect of autophagy on cell survival/proliferation,is also reported [Table 3].Thus,tumor necrosis factor-related apoptosis inducing ligand (TRAIL) induced autophagy and cell death in TPC1 cells,and the cell death effect of TRAIL was ameliorated byATG7siRNA,indicating anti-survival effect ofTRAIL-inducedautophagy,while in FRO cells,only cell death,not autophagy,was induced by TRAIL,but,similar to TPC1,TRAIL-induced cell death was impaired byATG7siRNA,indicating antisurvival effect of basal level of autophagy (NOTE -only LC3-II levels were determined in TRAIL-treated TPC1 cells.)[98].In another study,family with sequence similarity 129 member A (FAM129A) induced autophagy in parental PCCL3 cells,but inhibited RET/PTC-induced autophagy in RET/PTC-transfected PCCL3 and thyroid cancer cell lines PTC1 and FTC-236 (NOTE -mCherry-eGFP-LC3B produced yellow color in FAM129A-transfected PCCL3 cells,indicating FAM129A-inhibition,not induction,of autophagy.)[99].
(4) Possible reasons for inconsistent data
Although the reason(s) for these discrepant data are at present unclear,possible explanations are discussed below.
Obviously,autophagy is originally recognized as a pro-survival stress response,so that it is not difficult to acknowledge the pro-survival effect of autophagy on cancer cells.Indeed autophagy in general inhibits apoptosis (and vice versa),albeit with some exceptions[19](see below),thereby autophagy inhibition increases cell death,and cell-survival effects of various autophagy inducers are abolished by autophagy inhibitors as summarized in Table 1.However,there are indeed some exceptions for autophagy-inhibition of apoptosis;for example,autophagy degrades pro-survival factor caveolin-1,activates caspase 8,and plays a role as scaffolds for apoptosis execution[19].Furthermore,autophagy is reported to be a back-up mechanism for death of cells defective in or resistant to apoptosis during development[100],and in cancer cells[101].Therefore,the consequence of modulation of autophagic activity may be dependent on the sensitivity/resistance of the cancer cells to apoptotic stimuli;apoptosis-resistant cells may be vulnerable to autophagy inducers.Autophagy is also known to modulate other types of cell death[19]which,however,have never been studied in the thyroid cells.
The degree of dependency on or addiction to autophagy in cancer cells may determine response to autophagy modulators[102].Autophagy dependency is reportedly controlled by p53 status,RAS/RAF family status,JAK/STAT/PI3K signaling pathways,and so on.In fact,BRAFV600Epromotes autophagy dependence in multiple tumors[37,102-104],presumably as do RAS and RET/PTC.Thus,thyroid cancers,most of them have mutant RAS/RAF or RET/PTC,likely depend on autophagy for their proliferation/survival.
There are also some lines of evidence supporting anti-survival/proliferative effect of autophagy.The autophagic machinery executes specific forms of cell death,called “autophagic cell death” or “autophagydependent cell death”[19,20,105].The mechanisms include:(1) over-eating due to excessive-consumption of cellular organelles and cytoplasmic contents;(2) excessive mitophagy leading depletion of mitochondria and consequent energy failure;and (3) autosis by activation of Na+/K+ATPase pump and changes in membrane osmolarity and ion transporter.In this regard,it may be worth noting here that Liet al.[73]mentioned that “although sustained and profound autophagy activation could promote cell death (autophagic cell death),drug-induced feedback autophagy activation is mostly gentle and has pro-survival ability especially in cancer cells”.It is also of interest that rosuvastatin (a statin) induced autophagy at lower concentrations,which shifted to apoptosis with higher concentrations,in BCPAP cells[106].More recently,dendrogenin A (DDA),which is a cholesterol-derived metabolite and a ligand of the liver X receptor and is reported to induce lethal autophagy in other cancer cells[107],was demonstrated to induce cell death/cell cycle arrest and re-differentiation in thyroid cancer cell lines,B-CPAP and 8505C,although the consequence of autophagy inhibition from DDA is not evaluated[108].
Autophagy also controls thyroid differentiation status.Thus,there is a significant association between LC3 puncta and RAI uptake capacity/solute carrier family 5 member 5 [SLC5A5,or sodium/iodine symporter (NIS)] expression/clinical response to RAI[54].Experimentally,some of autophagy inducers (all are digitalislike compounds) increased RAI uptake in at least one of three PTC or FTC cell lines (BCPAP,TPC1,and FTC133) and in both ATC lines (8505C and Cal-62) (NOTE -all the autophagy inducers examined did not do so in all the cell lines examined,so that this effect may be attributed to their distinct intrinsic nature,not their autophagy-inducing ability,of digitalis-like compounds and/or different cells used,and also whether this effect is abolished by an autophagy inhibitor or not should be addressed.)[109,110].
Keeping all these possibilities in mind (summarized in Figure 3),although currently researchers mainly focus on autophagy inhibitors as a therapeutic modality for thyroid cancer,excess autophagy should also be considered as another choice.
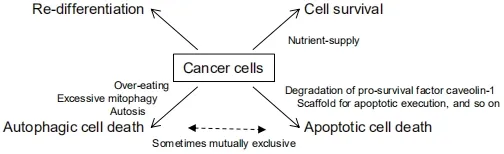
Figure 3.The consequences of autophagy activation in cancer cell phenotype/fate.Autophagy contributes cancer cell survival by providing nutrients,but conversely can induce apoptotic or autophagic cell death.Autophagy also re-differentiates cancer cell
Finally,of interest,intra-nuclear cytoplasmic inclusion,known as one of the diagnostic criteria for PTCs,is recently reported to be formed by nuclear invagination and contain autophagy-related proteins such as LC3,p62,ubiquitin,and cathepsin B/D,and also BRAFV600E[111],although its clinical significance remains to be elucidated.
CONCLUSION
The physiological role for autophagy in the thyroid is relatively clear that studies with genetically engineered mice show that basal level of autophagy is required for maintenance of cell homeostasis and survival,and elevated autophagy,in TSH-stimulated condition,is needed for providing building blocks for sufficient synthesis of proteins including TG.Regarding its pathological role in thyroid cancer initiation,there is some evidence indicating anti-tumorigenic effect of autophagy on cancer initiation in general,but so far nothing in thyroid cancer.Regarding thyroid cancer progression and autophagy-targeted treatment,the data are controversial,some reporting pro-survival/proliferative,but others showing anti-survival/proliferative and context-dependent effects of autophagy on thyroid cancer.Further studies are needed to elucidate the molecular mechanisms for the complex relationship between autophagy and its effect on thyroid cancer behaviors.Finally,precise monitoring of autophagic flux is highly critical for these studies.
DECLARATIONS
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was in part supported by the Japan Society for the Promotion of Science (KAKENHI Grant No.19K09028).
Conflicts of interest
The author declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2021.
 Journal of Cancer Metastasis and Treatment2021年1期
Journal of Cancer Metastasis and Treatment2021年1期
- Journal of Cancer Metastasis and Treatment的其它文章
- Metabolomics in cancer and cancer-associated inflammatory cells
- Bulbine frutescens phytochemicals as novel ABCtransporter inhibitor:a molecular docking and molecular dynamics simulation study
- The use of liquid biopsy in early breast cancer:clinical evidence and future perspectives
- Epigenetic control of autophagy in women’s tumors:role of non-coding RNAs
- Multiplexed bioluminescence imaging of cancer cell response to hypoxia and inflammation in the caudalartery injection model of bone metastasis during zoledronic acid treatment