
基于多位点序列分型方法的于桥水库微囊藻遗传多样性分析
2021-07-17 03:25乔之怡
水生生物学报 2021年4期
郑 涛 曹 琪 朱 梅 乔之怡
(天津农学院水产学院, 天津市水产生态与养殖重点实验室, 天津 300384)
随着全球水体富营养化水平的逐渐提高, 由于藻类大量繁殖造成的蓝藻水华问题成为淡水生态系统的共同威胁。全世界大约85%的淡水蓝藻水华由微囊藻占优势[1], 而且还有加重的趋势, 因为近年来的全球变暖可能会加剧不同水域中微囊藻暴发的频率、持续时间和泛滥程度[2]。
目前, 世界上已发现的微囊藻共有50多种。传统形态学分类根据微囊藻的群体形态、群体外胶被特征、细胞排列方式和气囊特点等来分种。然而, 在不同条件下微囊藻的群体形态可能会有很大变化[3], 这种现象使微囊藻的分类变得困难, 因此,也有学者认为现有微囊藻仅存一种, 即将铜绿微囊藻(Microcystis aeruginosa)、挪氏微囊藻(Microcystis novacekii)、鱼害微囊藻(Microcystisichthyoblabe)、绿色微囊藻(Microcystisviridis)和惠氏微囊藻(Microcystiswesenbergii)统称为铜绿微囊藻[4]。微囊藻可产生多种有害次生代谢物, 其中最为常见的微囊藻毒素(MCs), 可对肝、肠、脑、肾、肺、心和生殖系统造成不可逆损伤[5,6]。微囊藻形态多变, 仅靠形态难以对有毒无毒藻株进行区分。也正由于微囊藻的广泛分布和其带来危害的严重性, 微囊藻的遗传特性得到了广泛的研究[7,8]。
诸多证据表明地理隔离是微生物遗传分化的重要力量[9—11]。一般认为水系隔离较强就有更高的遗传多样性, 但是Zhu等[12]应用ITS作为遗传标记发现, 在对一个孤立的和超富营养化池塘进行的连续监测中, 微囊藻种群内具有较高的遗传多样性。Huo等[7]应用高通量测序的方法也在中国北方某水库中发现微囊藻的多样性极高, 这一结果证实了同一水系中微囊藻的多样性也会有所不同。所以利用多样性较高的分子遗传标记广泛对各水系进行研究, 才有可能较清楚的解释微囊藻变异与地理因素的关系, 可是相关研究较少报道, 所以地理因素对微囊藻遗传多样性的影响变得更加深远复杂[13]。这需要用更合适的方式进一步评估微囊藻种群之间相似性及分析遗传多样性形成中进化力量的作用。
采用高通量测序方法研究微囊藻的遗传多样性与环境因素之间的关系已被证明是可靠的[7]。这种方法可以获取较多的信息, 但数据分析工作专业性强, 消耗时间长, 难以在短时间内进行种群结构和多样性研究。单基因序列多样性分析技术也被应用于微囊藻上, 研究发现重组能在很大程度上促进微囊藻的自然遗传多样性, 并强调了基因交换对微囊藻种群遗传结构的潜在重要性[14], 如常用的序列有ITS[15]和cpcBA-IGS[16]等。目前, 与全基因组测序一样, 多位点序列分型的方法常被用在对地理距离较远的种群中, 以充分评价地理因素在微囊藻精细遗传结构发育中的作用, 其中管家基因已被证明是有效的遗传标记, 这也使推断出决定和限制微囊藻种内遗传多样性的其他进化因素成为可能[17,18]。
多位点序列分型(Multilocus Sequence Typing,MLST)是近年来分子生物学研究中发展较快的方法之一。MLST技术最初是在1998年为脑膜炎奈瑟菌(Neisseria itidis)研发的, 目的是解决不同实验室之间的分子分类重复率低的问题[19]。MLST一般检测6—10个管家基因中400—600 bp的核苷酸序列。每个位点的序列根据被发现的时间分配一个等位基因数, 每个菌株的等位基因数按指定的顺序排列为其等位谱, 即菌株的序列类型(Sequence type,ST)。因此, 每个ST代表一组核苷酸序列信息。通过比较ST, 可以发现菌株之间的相关性, 即亲缘关系较近的菌株有相同的ST, 或者只有少数不同的ST基因位点, 而亲缘关系较远的菌株则至少有3个或3个以上不同的ST基因位点。由于其高分辨率,在国际菌株研究中经常被用来确定流行病菌抗原的来源[20]。大量研究表明, MLST是一种介于在单基因和全基因组测序之间有足够的遗传信息且较为简便快速的分析方法[21,22]。目前缺乏更精细的微囊藻种内的分类依据, 从DNA分子多样性有可能筛选到合适的分类标记, MLST可以有更精细的分辨率, 有成为分类分子标记的潜在可能性。但目前为止, 仅有少数学者将MLST应用于蓝藻遗传多样性的研究, 同时在他们的研究中提出应用MLST研究微囊藻应建立更加丰富的数据库[17,18]。
于桥水库曾是天津唯一的饮用水源地。近年来, 微囊藻在夏季的库中占绝对优势且组成复杂[23]。研究其种群结构具有对微囊藻藻华的发生进行监测和控制等特殊的生态意义[7]。虽然之前于桥的研究已经包括了水库的水化指标[24,25]、蓝藻生态模型建立[26]和其他内容。但这些研究都是基于传统的计数和理化指标的测定, 而微囊藻群体形态特征高度可变, 甚至有学者对微囊藻属种水平的分类提出异议[1]。
本研究利用多位点序列分型分析技术对天津于桥水库的10株微囊藻株的7个管家基因进行分析,并与20个鄱阳湖序列型和237个日本湖泊序列型(STs)进行比较, 在研究我国北方水库型水体中微囊藻的基因型组成变化和遗传多样性的基础上, 进一步探究地理隔离与种内进化在与微囊藻MLST多样性的关系。
1 材料与方法
1.1 微囊藻藻株的分离和培养
2018年7月20日分别在于桥水库的放水洞、库中、库东、库西、库南、库北和峰山7个站位取样50 mL等比例混合后, 24h内带回实验室用经典毛细管法, 在灭菌CT[27]培养基中单克隆培养微囊藻藻株, 培养温度25℃, 光照强度25 μmol protons/(s·m2),昼夜比12h∶12h。微囊藻藻株纯化后移至含有无菌CT培养基的5 mL玻璃试管中。
1.2 16S rDNA、cpcBA和mcyE基因的检测
毒素基因的检测采用mcyE片段, 退火温度为55℃, 其余条件与管家基因相同[18]。16S rDNA基因PCR程序为: 94℃预变性5min, 94℃变性30s,54℃退火30s, 72℃延伸1min 30s, 此3个过程为30个循环, 最后72℃终延伸5min。cpcBA基因PCR程序为: 94℃预变性2min, 94℃变性30s, 55℃退火30s,72℃延伸30s, 此3个过程为35个循环, 最后72℃终延伸5min。基因检测的引物序列如表1所示。
1.3 MLST基因的检测
表1显示本研究使用的7个MLST管家基因, 包括ftsZ、glnA、gltX、gyrB、pgi、recA和tpi[17,18]。每个基因座都为单拷贝形式存在, 使用全细胞PCR对目的片段进行扩增。PCR扩增体系在柳满森等[17]的实验基础上做出改变, 将总体积改为25 μL,反应条件为: 94℃预变性3min, 94℃变性60s, 60℃退火30s, 72℃延伸30s, 此3个过程为35个循环, 最后72℃延伸5min。PCR产物经1.5%琼脂糖凝胶电泳检测, 然后经TIANquick Maxi Purification Kit(Tiangen)纯化DNA, 并用pEASY-T1 cloning kits(TransGen)回收, 之后挑取单克隆进行测序, 利用N C B I下载相关基因序列, 构建局部基因库。MLST对每个位点及不同的等位基因都分配了一个随机数, 7个等位基因的唯一组合明确定义了一个藻株类型序列(ST)。
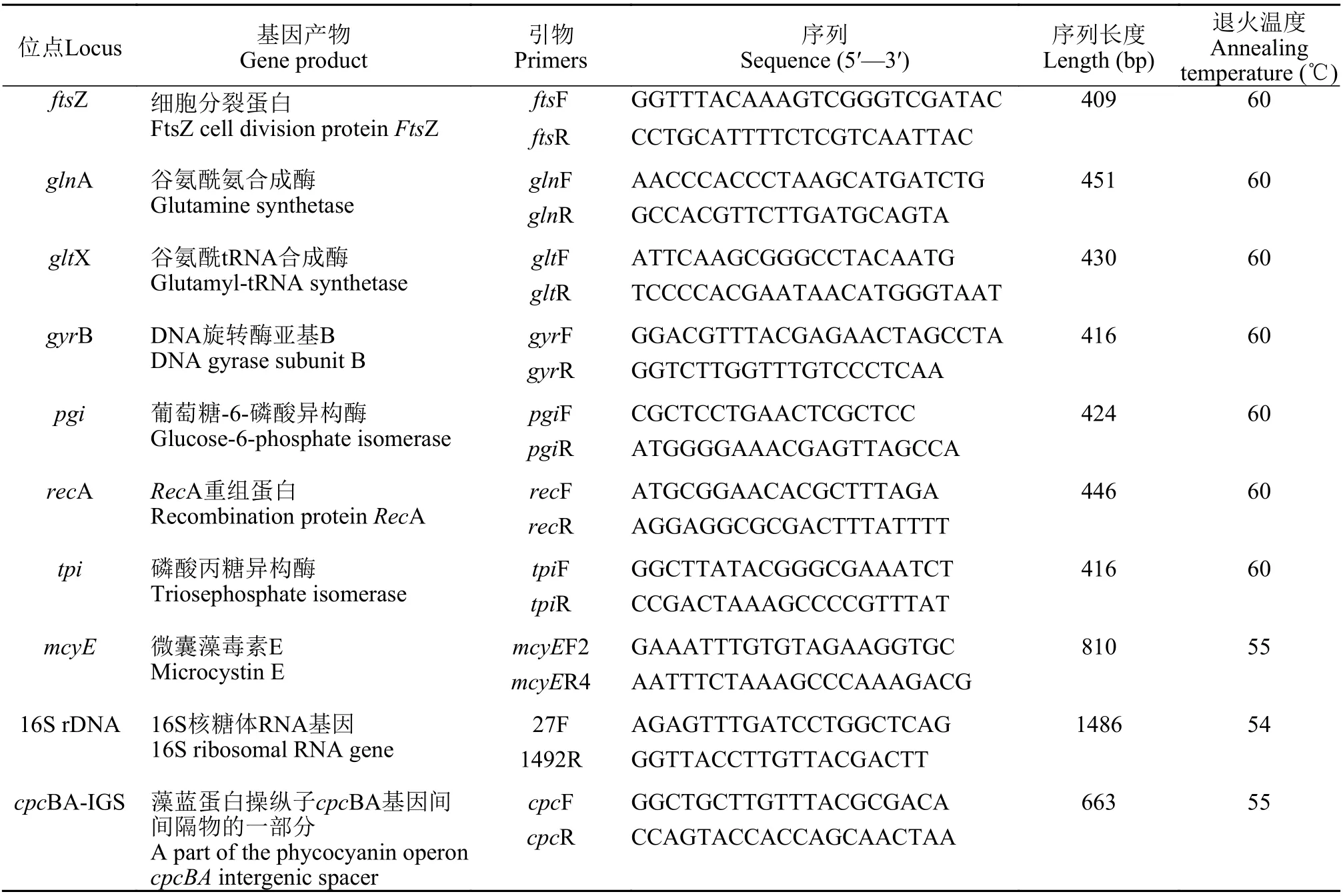
表1 分别用于MLST和mcyE基因、16S rDNA基因和cpcBA基因的PCR引物序列Tab. 1 PCR primer sequence used in MLST and PCR primer sequence of mcyE gene, 16S rRNA gene and cpcBA gene
1.4 七个管家基因的重组分析
从GenBank中选取MicrocystisPCC 7806作为参考亲本株系。使用Sim Plot 3.5进行潜在重组事件的相似性检测[28], 窗口长度为20 bp, 每步为20 bp。将统计数据导出后, 在Excel中对数据进行作图分析。
1.5 种群遗传和系统发育分析
采用DNAsp[29]分析DNA遗传多样性指数, 具体计算公式如下:,基于Tajima’sD和Fu and Li’sD[30]检验核苷多样性。系统发育分析采用Fasconcat-g软件, 按照ftsZ-glnAgltX-gyrB-pgi-recA-tpi的序列顺序连接7个管家基因, 然后在MEGA X软件中对连接顺序进行对齐,使用jModeltest对模型进行推断, 计算参数。用IQTREE建立最大似然树。
2 结果
2.1 基于MLST的序列差异和系统分析
本研究从10株微囊藻中分别扩增得到得到了7个MLST基因片段, 将本实验得到的MLST测序结果与前文提到的构建的MLST本地基因库进行比较, 得到10个新的ST型(ST258-ST267)。如表2所示, 每个ST对应唯一的微囊藻藻株。
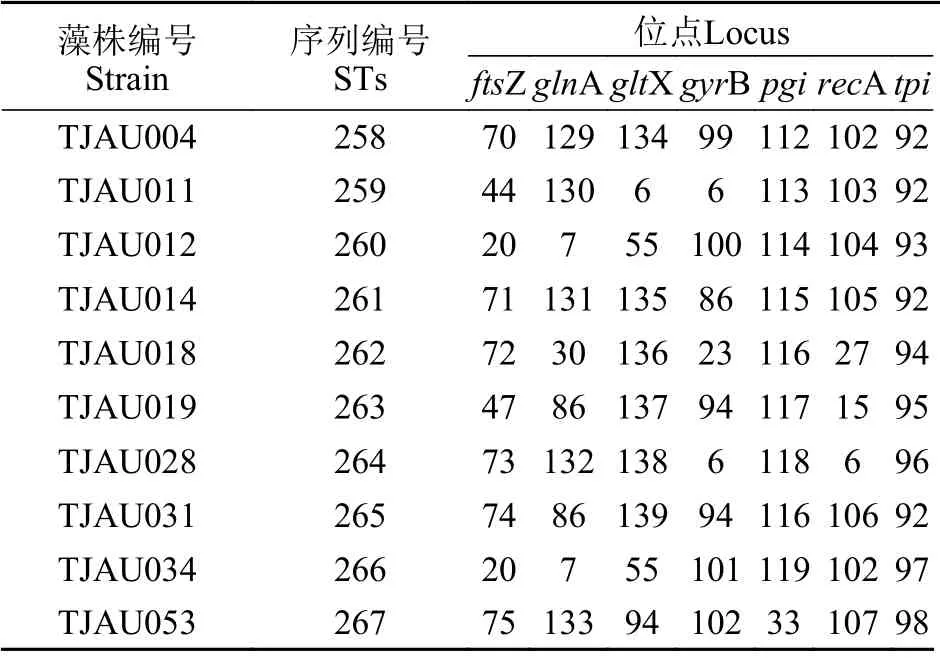
表2 于桥水库10株微囊藻的MLST序列及相应的等位基因数目Tab. 2 MLST sequences and numbers of 10 strains of Microcystis from Yuqiao Reservoir
2.2 mcyE基因检测及16S rDNA和cpcBA基因的系统分析
本研究藻株的mcyE基因检测结果均为阴性,说明它们产出微囊藻毒素的几率较低。分别将得到的10株微囊藻的16S rDNA基因和cpcBA基因片段添加进GenBank数据库中, 构建系统发育树(图1)。2个系统发育树的拓扑结构均显示本研究的10株藻株均位于微囊藻属的分支上, 证明它们均为微囊藻属。图1A为16S rDNA系统发育树, 将10株藻分为3组, TJAU1单独一组, TJAU2、3、5、9、10聚在一起, 同时TJAU4、6、7、8聚在一起; 在图1B中, 除藻株TJAU8外, 其他9藻株紧密聚集在一起。
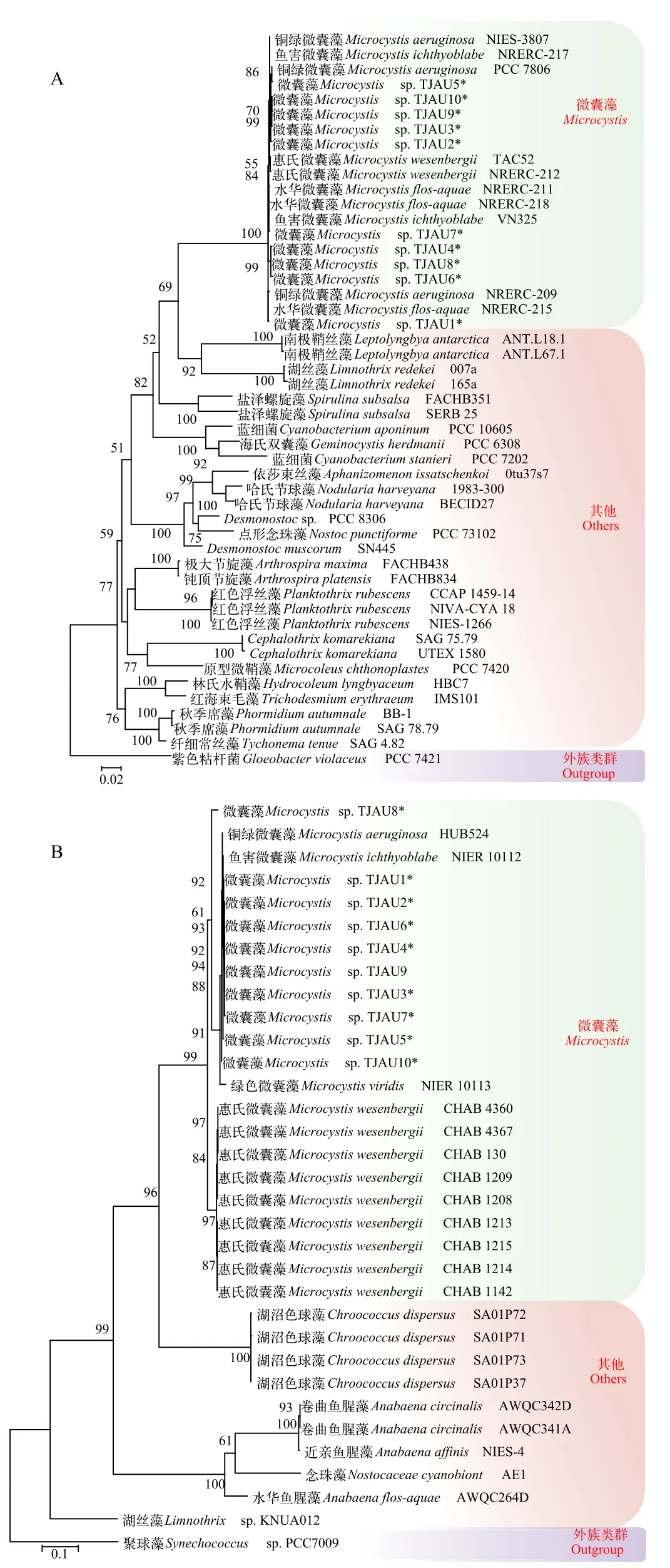
图1 基于蓝藻16S rDNA和cpcBA的系统发育树Fig. 1 Phylogenetic tree based on 16S rDNA and cpcBA of Cyanobacteria
2.3 基因重组分析
以标准微囊藻株PCC 7806作为参考株系, 使用Simplot重组分析工具对得到的7组MLST基因片段进行了基因重组分析, 如图2所示:ftsZ的序列相似性达到100%,recA的序列相似性则多在95%以上,且突变频率均匀, 说明ftsZ与recA序列不存在重组的可能。glnA、gltX、gyrB、pgi与tpi序列检测显示, 序列相似性多在95%以下且检测到不同程度的重组信号。
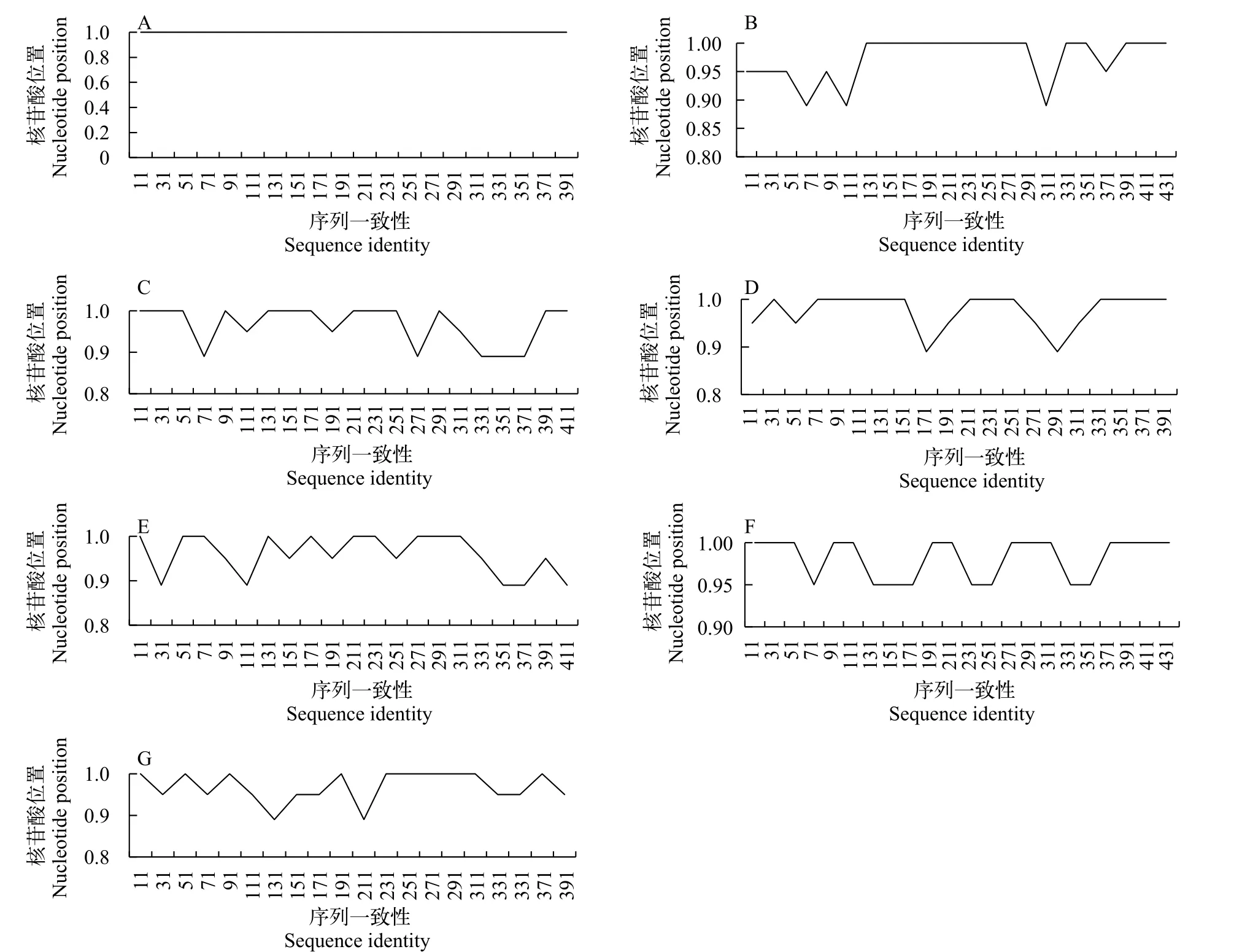
图2 重组的检测Fig. 2 Detection of recombination
2.4 基因多样性
在删除MLST序列两端的多余序列后, 在任何位点的序列中都没有发现插入或删除, 因此可以对序列进行对齐。DNAsp软件计算的遗传多样性指数如表3所示。本研究从10株藻中获得了10个STs。等位基因的数量从gyrB的14个到pgi的39个不等。总核苷酸多样性平均值为1.883%, 其中gltX最高为2.616%,tpi最低为1.106%。本试验所测于桥水库的MLST基因片段长度在409—451 bp, 平均遗传多样性H=0.938, Tajima’sD、Fu、Li’sD*均未拒绝中性遗传多样性的原假设。
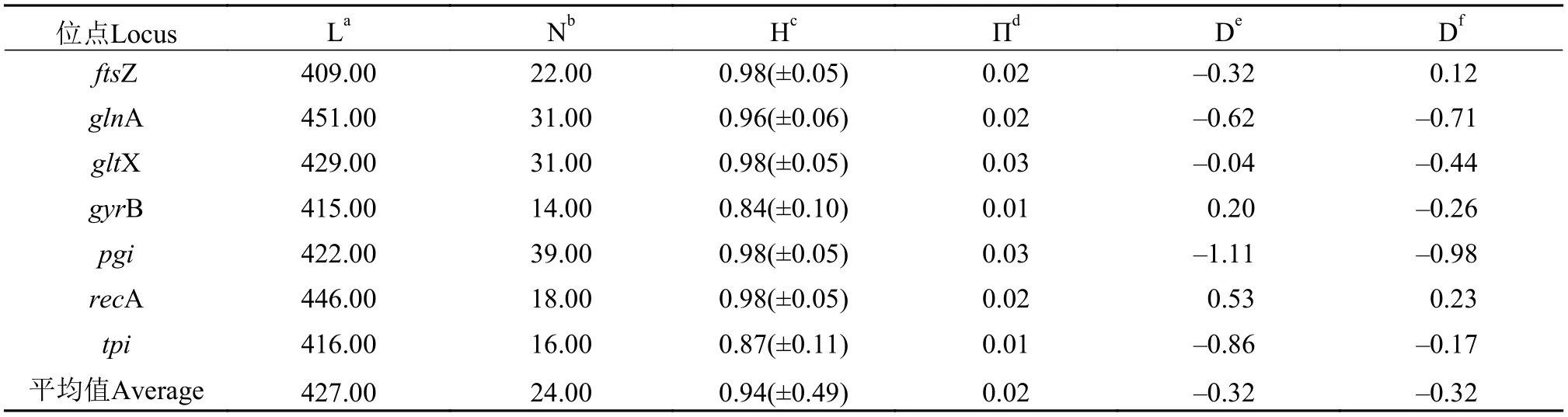
表3 微囊藻藻株基因多样性指数Tab. 3 Genetic diversity index of Microcystis strains
2.5 进化分析
为了缓冲重组对进化结果的影响, 将总长度为2992 bp的7个MLST序列串联起来[31]。采用JModeltest软件进行分层似然比检验和多位点序列进化分析发现, 最优模型为GTR + F + G4, 四个碱基的频率分别为0.2842、0.2267、0.245和0.2441。替代模型的速率矩阵为R(d)[C–G]=1.8512, R(e)[C–T]=6.1697, R(f) =1.0000。以于桥水库10个STs、鄱阳湖20个STs和日本湖泊237个STs为基础的MLST系统发育树如图3所示。ST1-ST237为日本湖泊的序列类型, ST238-ST257为鄱阳湖的序列类型, ST258-ST267为于桥水库的序列类型。结果表明, 藻株之间的亲缘关系得到了很好的解释, 系统发育树形成明显分枝, 其中于桥水库的微囊藻全部聚集于一个簇。

图3 利用于桥水库(●)、鄱阳湖(▲)和日本湖泊微囊藻属的STs进行系统发育分析Fig. 3 Phylogenetic analysis was performed on STs of Microcystis strains from Yuqiao Reservoir (●),Poyang Lake (▲) and Japanese lakes
3 讨论
应用基于7个管家基因的多位点序列分型技术(MLST)对分离自于桥水库的10株微囊藻的序列类型进行分析, 结果发现, 在所有试验的藻株中未发现完全相同的基因型, 与前人关于STs很少同时出现在2个以上的位置的研究结果相似[17,18]。这表明了于桥水库微囊藻存在广泛的遗传多样性。
本研究采取无差别随机取样方式获取了10株微囊藻藻株, PCR检测产微囊藻毒素mcyE基因, 结果均为阴性。由此推测在于桥水库中产毒素藻株总体比例偏低。这与王捷等[32]在我国北方另一水体, 汾河太原区段的研究中提出的微囊藻产毒素能力偏弱是一致的。有研究揭示许多种类的细菌具有降解微囊藻毒素的能力[33], 微囊藻产毒株的竞争能力在北方水体中可能受到了水温和气候, 甚至水体细菌等因素的影响而整体偏弱。
基于分子生物学的微囊藻种群时空分布调查已经成为了解微囊藻群体遗传规律和追踪水华发生过程的常规手段[34]。统计结果显示, 于桥水库微囊藻具有较高的遗传多样性。平均基因多样性H=0.938, 略低于鄱阳湖的微囊藻基因多样性(H=0.986)[17]和日本湖泊的微囊藻基因多样性(H=0.951)[18], 但远远高于大肠杆菌(H=0.47)[19]和枯草芽孢杆菌的基因多样性(H=0.44)[35]。对微囊藻的高遗传多样性的一种解释是基因可能存在重组, 通过基因重组, 可产生大量的遗传变异, 遗传多样性区域的增加与重组率的增加相关[36]。本研究结果显示7个管家基因中有5个可能发生了重组。另一种对高遗传多样性的解释是, 微囊藻包含几个生态上不同的种群或生态型, 这与刘满森等[17]的研究结果相近。考虑到生态分化与遗传分化的关系, 一个物种如果有更多的生态型, 其遗传多样性就会保持得更高[11]。从于桥水库中已分离出10种不同的STs,表明于桥水库水体环境条件可能并不稳定[37,38], 对于微生物来说, 四季水环境的波动造成了精细的生态位差异, 而水体中共存的微囊藻基因型是环境波动条件下的竞争结果, 这与孙千千等[39]得出的温度是驱动季节演替的主要因素和风引起的迁移是影响微囊藻群落空间分布的重要因素的结果一致。
ML树显示, 在大的空间尺度上于桥水库的序列类型聚在一起, 这可能是由于于桥水库中藻株种源结构较为一致或是生态上不同的种群经过反复的选择性清除, 从而造成基因组中多数基因位点高度一致[11]。结果还显示, 基于16S基因构建的系统发育树中TJAU1与其他8个藻株相距较远。基于cpcBA基因构建的系统发育树中TJAU8距离其他9个藻株较远。有证据表明, 一些微生物种群可能保持了大量的精细基因型和表型多样性, 因此这2株藻可能进化成为不同的基因亚型[40]。结合中性检验的结果, 说明遗传漂变可能是同一位点遗传多样性高的主要原因。虽然基于16S rDNA与cpcBA基因构建的进化树都表明本实验所用的10株藻株为微囊藻属, 然而, 在本实验中cpcBA与16S rRNA的拓扑结构并不完全一致, 这也表明虽然这些基因相对保守, 但系统发育不一致仍然存在, 不能排除同源重组增强了微囊藻的种群优势形成[41]。在基于16S rDNA和cpcBA基因的系统发育拓扑结构中,并没有显示出大空间尺度上明显的地理分布格局,这说明地理隔离可能不是影响微囊藻种群遗传的最重要因素。MLST构建的进化树对于桥地区的序列进行了区分, 表明在小生境内产生了一些具有小区域特征的变异, 这与Huo等[7]的研究结果相似。于桥水库微囊藻类遗传多样性高, 可能是由于微囊藻包含多种不同的生态类型, 遗传差异往往伴随着生境的差异。如果一个物种存在更多的生态类型,那么该物种将保持更高的遗传多样性, 随着时间的推移, 每个生态型都有其基于基因序列的分支[42]。在微囊藻的系统发育树上观察到许多不同的分支,这表明每个分支可能代表一个独特的生态型。在后续构建更庞大的MLST数据库的研究中还需要与具体藻株所处环境进行分析, 结合藻类生理研究对北方水体中微囊藻广泛的遗传多样性形成机制进行进一步的探讨。
4 结论
结果表明, 于桥水库存在多种不同的微囊藻的基因型。本研究认为中国北方地区微囊藻的遗传多样性较大, 这在以往的研究中很少受到关注。将单基因分析与MLST方法进行比较, 可以发现单基因系统发育拓扑结构是不完整的, 这说明MLST可以提供更详细的信息。本文推测, 于桥水库微囊藻受遗传漂变和小生境选择的共同影响在种群发展过程中发生了一些基因的亚型分化。后续对微囊藻MLST的研究中需要更加关注环境因素的潜在影响。
猜你喜欢
区域治理(2022年40期)2022-11-27
中南药学(2022年8期)2022-11-19
今日农业(2022年15期)2022-09-20
湖南电力(2021年1期)2021-04-13
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
江苏农业学报(2019年1期)2019-09-10
红土地(2018年7期)2018-09-26
中国环境科学(2017年5期)2017-05-23
