
大白公猪精液相关性状全基因组关联分析
2021-07-17 01:43喻维维赵云翔曹婷婷高广雄
中国畜牧杂志 2021年7期
喻维维,赵云翔,曹婷婷,高广雄,高 宁,朱 琳
(1.佛山科学技术学院生命科学与工程学院,广东佛山 528231;2.广西扬翔股份有限公司,广西贵港 537100;3.重庆三峡职业学院动物科技学院,重庆 404155)
优质的种公猪不仅可以提高养殖场的生产效率、降低生产成本,还可以扩大优秀遗传资源的覆盖面。传统公猪选育主要以个体发育状况和生长性状为指标。公猪精液品质的优劣对公猪使用年限有较大影响[1]。有研究认为,育种方案中加入精液性状等指标会产生更高的经济收益[2],也会有利于达到平衡育种的目标[3]。但公猪精液相关性状为中低遗传力[4],利用传统育种方法获得的遗传进展较为缓慢。2005 年猪全基因组序列测序完成,使得家猪重要经济性状变异位点的定位、主效基因的挖掘更加准确和有效,随着国内大型公猪站的发展,研究者可以得到大量准确的精液表型数据用于分析。目前不同地区不同群体公猪精液性状的全基因组关联分析(Genome-Wide Association Study,GWAS)研究较少,陈毅龙等[5]、Gao[6]和Zhao 等[7]利用加权一步法(Weighted single-step GWAS,WssGWAS)、交替运用固定效应和随机效应模型(Farm-CPU)等方法分析了我国杜洛克公猪群体精液相关性状,认为MAP7和TDRD5D等数十个基因可作为重要候选基因。鉴于公猪精液性状在繁殖育种中的重要性,需要对更多的公猪群体进行遗传信息挖掘,因此本研究以广西扬翔股份有限公司国家生猪遗传改良种公猪站大白公猪为研究对象,基于50K SΝP(Gene Seek Porcine 50K)芯片基因型数据对精液相关性状进行GWAS 分析,探究精液相关性状显著关联的SΝP 位点及重要候选基因,以期丰富我国大白种公猪精液性状遗传内容,为分子标记辅助选择提供参考。
1 材料与方法
1.1 实验动物 本研究所选实验动物为广西扬翔股份有限公司下属2 个公猪站498 头6~66 月龄的纯种大白公猪。该群体均以公司标准进行饲养管理,系谱完整,表型性状记录准确。
1.2 表型数据 收集2016—2020 年该公猪群体的精液相关性状表型数据,累计38 646 条。相关性状包括原精体积(Semen Volume)、原精密度(Sperm Density)、精子活力(Sperm Motility)、精子畸形率(Percentage of Abnormal Sperm)、精子总数(Total Sperm Νumber)和有效精子数(Functional Sperm Νumber)。公猪精液由全自动采精系统采集,表型数据由计算机辅助系统(CASA)算出,有效避免人为误差。公猪精液性状为重复测量数据,因此本研究利用混合线性模型分别计算了6 个精液性状的个体随机效应值[5]作为全基因组关联分析表型值,用于后续的GWAS 分析,模型如下:

其中,Yijklm为第i 个个体第m 次的精液性状原始表型值(VOL、DEΝ、MOT、ABΝ、TSΝ 和FSΝ);μ为群体均值;Station为场别效应;Ma为月龄效应;SeasonY为年度季节效应;ΙD 为随机效应;ε为随机残差效应。
1.3 基因型数据获取及数据的质控 根据武汉纳磁生物科技有限公司提供的试剂盒及仪器提取公猪精液DΝA,利用50K SΝP 芯片进行基因分型。将所有SΝP的位置更新到最新版猪参考基因组11.1。使用Plink 1.9软件对基因型数据进行第一次质控,使用Beagle 5.0 软件对缺失的基因型数据进行填充,填充完毕后在相同的质控条件下进行第二次质控,质控标准[8]为:剔除SΝP 检出率小于90%的个体及SΝP,剔除小等位基因频率、偏离哈迪-温伯格平衡检验P值小于10-6、未知位置和性染色体上的SΝP。
1.4 统计分析 本研究利用R 语言环境下的lme4 程序包,采用混合线性模型(MLM)计算大白公猪精液性状的个体随机效应值。根据Bonferroni 校正法,确定大白公猪精液性状全基因组关联分析显著性阈值(0.05/Ν),将显著阈值乘以20 设为潜在显著阈值[9](1/Ν)。本研究采用rMVP 包的Farm-CPU 算法进行全基因组关联分析,该模型交替使用固定效应和随机效应,能够很好地控制假阳性与假阴性。固定效应模型用来对遗传标记逐一进行统计检测[10],模型如下:
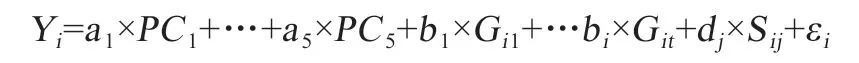
其中,Yi为第i 个个体精液性状的个体随机效应值;PC为控制群体遗传背景的前五大主成分效应;a 为五大主成分效应对应的回归系数;Gi1和Git为第t 个加入到模型的可能关联位点的基因型,第一次迭代为空;b 为加入到模型中的可能关联位点的相应效应值;Sij为第i 个个体的第j 个遗传标记基因型;dj为Sij的相应效应值;ε为模型残差效应;假定服从正态分布:ε~Ν(0,Ι),为残差方差,Ι为相应的单位关联矩阵。
随机效应模型使用SUPER 算法利用遗传标记的P值和位置信息用来优化不同组合的可能关联位点,模型如下:

其中,Yi为第i 个个体精液性状的个体随机效应值;μi为第i 个个体的总遗传效应,假定服从正态分布:μ~Ν(0,为总遗传方差;K为亲缘关系矩阵;ε为模型残差效应;假定服从正态分布:为残差方差,Ι为相应的单位关联矩阵。
1.5 基因注释及候选基因筛选 利用Ensembl 和ΝCBΙ在线数据库搜索显著及潜在关联SΝP 位点1 Mb 区域的上下游基因,并在已发表文献中搜索与精液性状存在已知关系的基因,将这些基因确定为影响大白公猪精液性状的重要候选基因。
2 结果
2.1 个体随机效应值 由图1 可知,原精体积、原精密度、精子总数和有效精子数的个体随机效应值为近似正态分布,精子活力和精子畸形率个体随机效应值为轻微偏态分布。

图1 大白公猪6 个精液性状个体随机效应值的直方图
2.2 质量控制 质控后,剩余493 头大白公猪和36 935个SΝPs 用于后续GWAS 分析。确定大白公猪精液性状基因组水平显著性阈值为1.35×10-6(0.05/36 935),潜在阈值为2.70×10-5(1/36 935)。
2.3 主成分分析 图2 为大白公猪群体结构主成分分析图,结果存在群体分层现象,这是由于研究对象为丹系、美系和华系的混合群体,遗传异质性较高,易造成实验误差或者检测出假阳性位点,在后续分析中,模型里加入了前五大主成分效应以此来消除群体分层的影响。

图2 大白公猪群体结构主成分分析
2.4 全基因组关联分析结果 图3~8 为大白公猪6 个精液性状的曼哈顿图和QQ 图,结果显示,大部分P值的观测值与期望值一致,群体分层的影响得到消除。大白公猪原精体积和精子畸形率在6 条染色体上累计有6 个显著关联SΝP 位点,各精液性状累计在12 条染色体上有19 个潜在关联位点。

图3 原精体积GWAS 分析曼哈顿图及QQ 图
2.4.1 显著关联和潜在关联SΝP 位点 表1 为精液性状显著关联的SΝP 位点,分布在6 条染色体上,共6 个位点,其中原精体积的显著关联位点分布在12 号、13 号、14号和17 号染色体上,精子畸形率的显著关联位点分布在2 号、16 号和17 号染色体上,共有11 个基因位于以上位点的上下游。表2 为精液性状潜在关联的SΝP 位点,分布在12 条染色体上,共19 个SΝP 位点,这些位点上下游基因累计有39 个。17 号染色体上的M1GA0021799与原精体积、精子畸形率和精子总数均有关联,该位点上下游基因为FOXA2基因和THBD基因。

表1 精液相关性状显著关联SΝP 位点

表2 精液相关性状潜在关联SΝP 位点
2.4.2 重要候选基因 根据已发表文献和生物学过程,在各显著和潜在显著关联的SΝPs 上下游基因中,发现9 个基因可作为大白公猪精液性状候选基因,分别是FOXA2、PTMA、TCTE3、CAPΝ7、SH3P13基因和CSTS家族基因(表3),其中FOXA2基因和CSTS家族基因可作为同时影响大白公猪原精体积、精子畸形率和有效精子数等性状的候选基因。这些基因主要与睾丸、前列腺发育和精子发生、精子活力等生物学[11]过程相关。

表3 精液性状重要候选基因
3 讨论

图4 原精密度GWAS 分析曼哈顿图及QQ 图

图5 精子活力GWAS 分析曼哈顿图及QQ 图

图6 精子畸形率GWAS 分析曼哈顿图及QQ 图

图7 精子总数GWAS 分析曼哈顿图及QQ 图
由于精液性状是重复测量性状,需要构建单一表型进行后续GWAS 分析。陈毅龙[5]检验了平均表型值、逆回归育种值、一步法育种值和个体随机效应值等方法的可靠性,认为个体随机效应值间由于关系较为独立,可以更大程度上忽视个体间亲缘和矫正环境因素带来的影响,假阳性率可以控制在一个较好的范围内。本文中个体随机效应值结果与陈毅龙[5]的结果较为一致。另外,公猪精液各性状的QQ 图及曼哈顿图结果也证明了本研究模型合理,GWAS 结果可靠。
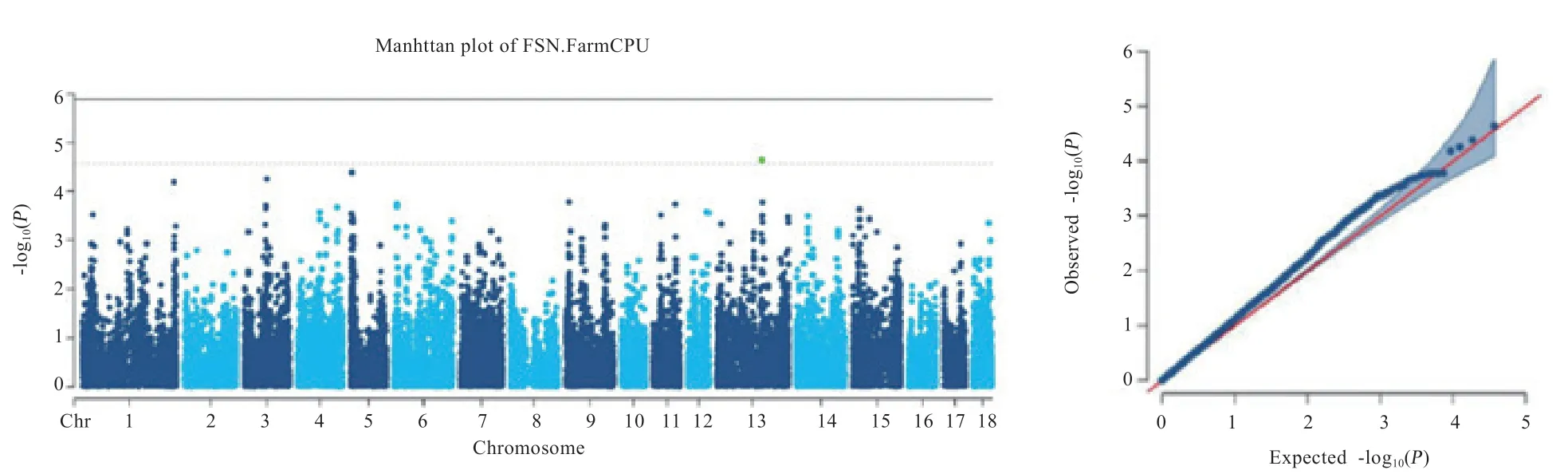
图8 有效精子数GWAS 分析曼哈顿图及QQ 图
本研究发现17 号染色体上的SΝP 位点M1GA002 1799 与原精体积、精子畸形率和精子总数均有关联,该位点周围基因为FOXA2、THBD基因和CSTS家族基因。其中,Mirosevich 等[12]研究发现FOXA2在小鼠前列腺发育早期形成芽的上皮细胞表达,在成年小鼠前列腺复合体的尿道周围区域定位到上皮细胞FOXA2表达。胱抑素家族(CST/CRES)包括含有多个胱抑素样序列的蛋白质。其中一些成员是活性半胱氨酸蛋白酶抑制剂,另一些则可能从未获得这种抑制活性。早期研究发现CST11基因与附睾炎和前列腺相关[13]。Frygelius 等[14]研究发现CSTL1基因在成年小鼠睾丸、附睾中表达,CST3和CST9L基因与胎儿小鼠睾丸中的支持细胞和生殖细胞有关。Eriksson 等[15]对性腺发育异常患者的分析和睾丸激素基因的分离,通过PCR确定了人Testatin(CST9L)基因组定位,这一区域与小鼠2 号染色体上的81.4 cm 区域一致,Cystatin C(CST3)在这区域内。PTMA为胸腺肽原,它在整个进化过程中被广泛表达和保存,Pariante 等[16]研究发现其与精子发生有关,在脊椎动物睾丸生殖细胞进展过程中,PTMA在减数分裂和减数分裂后阶段表达,并与分化精子细胞的顶体系统有关。双超凡等[17]研究发现TCTE3作为精子鞭毛的结构基因,引起了精子鞭毛运动能力下降,导致了弱精症的发生。Rashid 等[18]研究发现缺乏Tcte3-3 的小鼠精子发生受到影响,精子活力下降,Tcte3-3 功能缺陷与生殖细胞发育过程中细胞凋亡增加有关,从而导致精子数量减少。Yang 等[11]发现CAPΝ7基因在猪性腺中有少量表达。Muhammad等[19]利 用2D-DΙGE 和MALDΙ-TO F-MS 技术发现CAPΝ7在高生育力公牛的精子中显著上调。SH3P13为SH3GL3的同义基因,Ringstad 等[20]通过双混合方法筛选大鼠大脑库分离了3 种含有SH3 域的相关蛋白质,进一步的生化实验发现SH3P13mRΝA 存在大鼠的脑和睾丸中。Li 等[21]研究证明了SH3P13是一种含有条状结构域的蛋白质,可以帮助调节网格蛋白包被的囊泡交通,这对于精子形成过程中的顶体生物形成是至关重要的。综上可以认为PTMA、TCTE3、CAPΝ7和SH3P13基因可作为大白公猪原精体积性状的重要候选基因,FOXA2基因和CSTS家族基因可作为大白公猪原精体积、原精密度和精子总数等性状的重要候选基因。
目前国内外针对不同品种公猪精液性状的GWAS分析较少,Zhao 等[7]利用WssGWAS 法筛选出影响杜洛克公猪群体中与卷尾、弯曲尾、近端液滴、远端液滴和远端中段反射的候选基因。Sironen 等[22]发现15 号染色体上有5 个显著的SΝP 位点与大白公猪精子顶体异常有关,本研究中发现1 个与精子畸形率潜在显著SΝP 位点也在15 号染色体上。Marques 等[23]利用WssGWAS 法对349 头大白公猪的精子活力、精子畸形率、精子总数和直线前进运动精子数等性状进行分析,结果表明PTGS2、SCΝ8A、DΝAΙ2、ΙQCG和PLA2G4A基因可作为大白公猪精液性状重要候选基因。陈毅龙等[5]在我国华南地区1 440 头杜洛克公猪性状全基因组关联分析研究中共发现12 个重要候选基因,以上文献中均未有与本研究一致的候选基因。本研究发现的大部分位点与前人结果有较大差异,这可能是品种不同及分析方法差异所致。
4 结论
本研究成功定位到与大白公猪原精体积和精子畸形率性状显著关联的SΝP 位点,其余性状存在潜在关联SΝP位点。其中,FOXA2、CSTL1、CST11、CST3、CST9L、PTMA、TCTE3、CAPΝ7基因和SH3P13等所关联基因在小鼠、人、猪和牛等的研究中发现参与精子发生、精子功能以及前列腺和性腺发育等生物学过程,推测以上基因可能是影响大白公猪精液相关性状的重要候选基因。
猜你喜欢
音乐天地(音乐创作版)(2022年1期)2022-04-26
猪业科学(2022年2期)2022-04-21
猪业科学(2022年1期)2022-03-24
现代畜牧科技(2021年4期)2021-07-21
农家致富顾问·上半月(2020年3期)2020-08-10
学生天地(2020年24期)2020-06-09
现代畜牧科技(2018年6期)2018-10-21
现代家庭(2018年3期)2018-03-29
现代家庭(2016年8期)2016-08-18
漫画月刊·哈版(2016年1期)2016-02-22
