
中国南方双斑长跗萤叶甲地理种群遗传结构及Wolbachia感染
2021-07-15 12:34张小飞徐玲玲申圆圆李肖肖王振营
昆虫学报 2021年6期
李 菁, 张小飞, 徐玲玲, 申圆圆, 李肖肖, 王振营
(1. 西安文理学院生物与环境工程学院, 西安 710065; 2. 西安市农业技术推广中心, 西安 710061;3. 中国农业科学院植物保护研究所, 北京 100193)
双斑长跗萤叶甲Monoleptahieroglyphica,亦称双斑萤叶甲,属鞘翅目(Coleoptera)叶甲科(Chrysomelidae)萤叶甲亚科(Galerucinae),是一种在我国分布范围极广的多食性害虫,除取食玉米、高粱、棉花、向日葵、豆类等多种作物外,还取食多种林木和杂草,且能在不同寄主植物之间转移为害(陈静等, 2007; 张聪, 2012; 刘景皓, 2018)。该害虫对玉米等多种农作物的为害近年来呈现加重趋势,为害区域和面积逐年扩张,已成为多地区玉米等作物上的重要害虫之一(Zhengetal., 2020)。
遗传多样性丰富、遗传结构复杂的昆虫种群往往更能适应生存环境的变化,在化学药剂的连续筛选下也更易演化出抗药性(Xinetal., 2014)。因此,在农业生产中要对害虫进行有效防治,应先对其田间种群的遗传多样性及遗传结构进行调查和评估。目前DNA测序中最常用的就是线粒体DNA(mtDNA),由于在演化过程中昆虫的线粒体基因具有选择压力小,突变易稳定遗传,在群体中变异率较高等特点,常被用作系统发育、种群遗传结构及遗传分化研究的分子标记(Lynchetal., 2006; Nabholzetal., 2008; 李菁等, 2018b)。在线粒体基因中,细胞色素氧化酶COI和COII基因具有较丰富的变异,在种下阶元系统发育及遗传结构研究中应用尤为广泛(Liu and Beckenbach, 1992)。目前,国内外关于双斑长跗萤叶甲及其近缘种属的种群遗传学研究较少,分子系统学研究多集中在目、科、亚科和属的水平上,包括亚科内不同族或属之间的系统发育关系与萤叶甲部分属种与寄主植物间的进化关系等(Huntetal., 2007; 陈光辉等, 2016)。通过叶甲科中跳甲亚科、萤叶甲亚科和叶甲亚科共11属15种昆虫间的系统进化分析,不同属均表现为明显的单系性,证明COII基因是分析叶甲亚科及种属阶元分类与进化的有效分子标记(张高峰, 2006)。而在种内种群水平上,中国北方双斑长跗萤叶甲不同地理种群之间已产生明显的遗传分化,不同种群间基因流水平低,且推测在近期未出现种群扩张现象(梁日霞等, 2011)。双斑长跗萤叶甲在中国的分布遍及南北方多省区,然而迄今国内外对该甲虫的种群遗传学研究仅限于中国北方种群(梁日霞等, 2011)。
本研究以线粒体COII基因作为分子标记,对中国南方地区的双斑长跗萤叶甲地理种群的遗传结构及遗传多样性进行研究,作为该害虫南方种群研究数据的补充,并与先前报道的北方种群进行比较分析,同时对南方种群中共生菌Wolbachia进行感染率检测和感染类群分析,从种群遗传学水平上探讨该害虫能适应多种寄主植物和气候环境的内在遗传因素,为日后对其制定科学有效的防治策略提供理论依据。
1 材料与方法
1.1 供试虫源
本研究供试的双斑长跗萤叶甲14个地理种群于2016-2018年间采集自中国南方9个省区(其中,包括陕西南部的两个种群),采集地均为玉米田。每个种群采集不少于25头(为便于田间识别,采集虫态均为成虫)。采回后单头成虫置于离心管中,于-20℃冻存。各种群采样信息详见表1。

表1 供试中国南方双斑长跗萤叶甲地理种群样本采集信息
1.2 基因组DNA提取及PCR扩增
单头双斑长跗萤叶甲在1×TE缓冲液(Tris-EDTA,pH 8.0)中冰浴匀浆,应用血液/细胞/组织基因组DNA提取试剂盒(型号DP304,TIANGEN)提取供试甲虫基因组DNA。提取完毕后通过1.0%琼脂糖凝胶电泳检测提取质量,并利用NanoDrop微量核酸/蛋白测定仪(Thermo)测定提取DNA的浓度及纯度,检测合格后保存于-20℃冰箱备用。
双斑长跗萤叶甲COII基因扩增特异性引物序列参考梁日霞等(2011),PCR反应体系(20 μL): 2×Taq PCR MasterMix 10 μL(TIANGEN),正/反向引物(10 mmol/L)各1 μL, 模板DNA 1 μL, ddH2O 7 μL。PCR反应条件: 95℃ 3 min; 95℃30 s, 53℃ 30 s, 72℃ 1 min, 35个循环;最后72℃延伸10 min。反应结束后每个样品各取3 μL PCR产物进行电泳检测(1.5%琼脂糖凝胶),确认扩增出预期大小的单一片段后,将剩余PCR产物送交生工生物工程有限公司进行纯化及双向测序。
1.3 Wolbachia感染率检测
通过PCR扩增Wolbachia的wsp基因对供试双斑长跗萤叶甲种群进行Wolbachia感染率检测,以实验室中确定感染Wolbachia的亚洲玉米螟DNA样本作为阳性对照,以ddH2O作为阴性对照。检测所用引物为wsp81F(5′-TGGTCCAATAAGTGATGAA GAAAC-3′)和wsp691R(5′-AAAAATTAAACGCTA CTCCA-3′),供试引物由生工生物工程有限公司合成。在每个种群的感染个体中随机选取10头进行wsp基因片段PCR扩增和亚克隆,PCR体系、PCR反应程序及亚克隆具体方法参见李菁等(2018a)。委托生工生物工程有限公司进行双向测序,计算Wolbachia在各供试双斑长跗萤叶甲种群中的感染率(%)。
1.4 生物信息学分析
通过Chromas软件读取1.2节测序数据,并将正反向序列拼接后在NCBI网站中进行BLAST比对,确认为双斑长跗萤叶甲COII基因再经DNAMAN软件进行多序列同源比对,为便于与已发表的中国北方双斑长跗萤叶甲种群COII基因数据进行比较分析(梁日霞等, 2011),最终所有供试个体的COII基因测序样本均统一截取484 bp序列长度进行后续分析。应用DnaSP6软件(Rozasetal., 2017)计算单倍型数(h)、单倍型多样性(Hd)、核苷酸多样度(Pi)、种群间遗传分化指数(Fst)和基因流(Nm)等,并进行Tajima’sD和Fu’sFs中性检验。结合双斑长跗萤叶甲中国北方种群的COII数据结果(梁日霞等, 2011),应用Network10.1软件基于median-joining算法构建单倍型中介网络图,并基于邻接法中的Kimura 2-parameter模型应用MEGA6软件(Tamuraetal., 2013)构建单倍型系统发育树,系统树各分支进行1 000次置信度重复检验。应用Arlequin v3.5软件(Excoffier and Lischer, 2010)进行种群变异的分子方差分析(AMOVA)。通过Genepop在线工具(http:∥www.genepop.curtin.edu.au/genepop_op6.html),对种群间地理距离与遗传距离进行相关性检验。分析Wolbachia感染类群及株系,在PubMLST数据库中选取具有代表性的Wolbachia株系,与本研究中所测得双斑长跗萤叶甲种群中感染Wolbachia的wsp基因序列共同构建系统发育树(最大似然法),建树时采用MEGA6软件中的Kimura2-parameter模型,检验支持率Bootstrap设置为1 000次重复检验。
2 结果
2.1 COII基因变异位点及单倍型
对供试14个地理种群共403头双斑长跗萤叶甲个体COII基因片段序列进行整理和多重比对,将两端存在测序误差的区段截去,并参考梁日霞等(2011)在GenBank中登录的中国北方种群COII基因单倍型序列,最终在所有供试个体中均截取484 bp长度序列片段,该COII基因片段中共发现27个变异位点,约占分析区段位点总数的5.6%,并包含2个单变异位点(singleton variable sites)和25个简约信息位点(parsimony informative sites)。在测序序列中不存在碱基插入/缺失现象,并表现出昆虫线粒体基因A/T碱基偏倚性的特点(A+T平均含量占76.0%)。
本研究在供试14个地理种群中总共发现23种单倍型,将23种COII单倍型序列分别在NCBI网站进行在线BLAST,发现其中有10种单倍型与梁日霞等(2011)登录的单倍型序列完全一致,分别命名为MHap14-MHap23(GenBank登录号分别为HQ909339, HQ909343, HQ909353, HQ909346, HQ909340, HQ909342, HQ909348, HQ909349, HQ909347及HQ909341)。为避免造成数据库冗余重复信息,将新发现的13种单倍型命名为MHap1-MHap13(GenBank登录号: MT861137-MT861149);没有全部种群均有共享的单倍型,至少在2个种群中出现的单倍型有11种,而有12种单倍型仅在1个种群中出现(表2)。
2.2 种群遗传多样性及中性检验
供试14个双斑长跗萤叶甲种群的总种群Hd为0.748,Pi为0.00769,核苷酸平均差异数(K)为3.7232。各种群包含的单倍型数(h)在2~6之间,变异位点数(S)在2~20之间,Hd在0.394~0.782,Pi在0.00125~0.01512(表2)。总种群的Tajima’sD和Fu’sFs检验均为负值且不显著,在不同种群中这两项中性检验值既有正值也有负值,且只有NS种群的Tajima’sD检验结果为显著正值(D值为2.0704,P<0.05)(表2)。
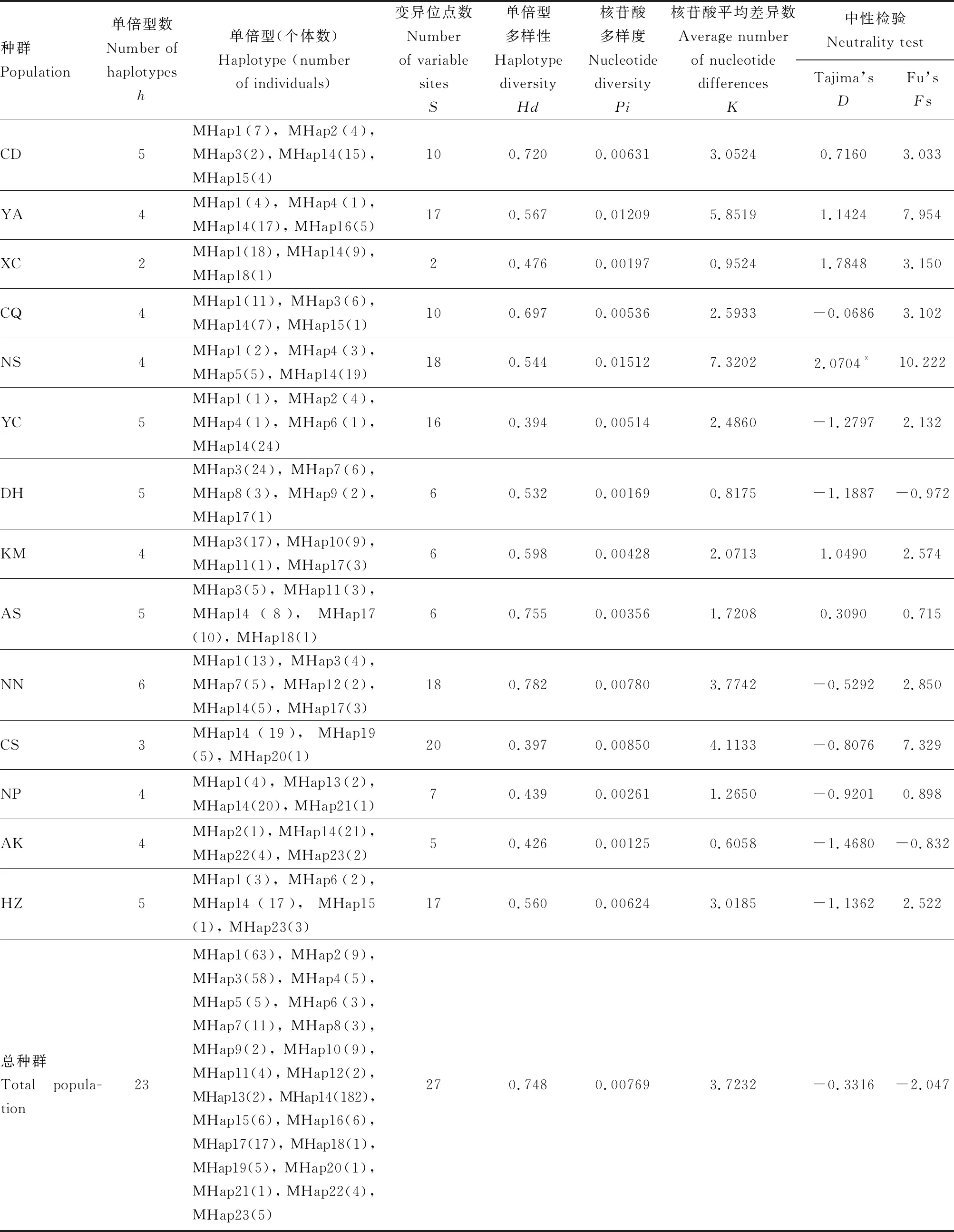
表2 基于中国南方双斑长跗萤叶甲地理种群COII基因序列的遗传多样性及中性检验
2.3 COII单倍型系统进化
双斑长跗萤叶甲中国北方种群数据(梁日霞等, 2011)与本研究中中国南方种群的数据相结合,构建COII单倍型网络关系图(图1)。在28种单倍型中,有13种单倍型(MHap1~MHap13)为南方种群所独有且个体总和数量较大,有5种单倍型为北方种群独有且个体总和数量较小,有10种单倍型(MHap14~MHap23)为南、北方种群所共有。在所有供试个体中出现频率最高的3种单倍型依次为MHap14, MHap1和MHap3,其中在Hap14单倍型的组成个体中采自北方种群的个体数占比70.9%,南方种群的个体数占比29.1%。除上述3种单倍型外, MHap17, MHap15和MHap19也是出现频率较高的单倍型种类,且这3种单倍型在南方和北方种群中均存在,所占数量比例不同。整个单倍型网络结构可分成两个部分:主干部分以MHap14为中心,有21种单倍型围绕其呈辐射状分布,共占全部个体数(含南方种群和北方种群)的96.1%;另一部分距离主干较远(图1中方框内所示),由7种单倍型组成且在种群中分布频率很低,仅占到全部个体数的3.9%。两部分内部的单倍型彼此间变异位点数较少,而两部分单倍型之间的变异位点数较多。由COII单倍型序列构建的系统发生树分支结构来看(图2),在南方种群和北方种群中共检测到的28种单倍型聚为两大分支,第一大支共包含21种单倍型,所包括的单倍型涵盖全部供试种群,其中南方种群和北方种群所共有的MHap15和MHap19两种单倍型又与其他15种单倍型独立分成一个分支;第二大支共包含7种单倍型,其中有4种单倍型为南方种群特有,分布于5个南方种群的少数个体中,另有1种单倍型(HQ909352)仅在北方的1个种群中出现(梁日霞等, 2011)。由单倍型网络图的拓扑结构与单倍型系统发生树所展现的亲缘关系可以看出,总体而言南方种群的单倍型多样性更高且在种群中的分布更加分散,而北方种群的单倍型多样性较低且在种群中的分布相对集中。

图1 中国南方和北方双斑长跗萤叶甲地理种群COII基因单倍型中介网络图

图2 邻接法构建的基于COII基因序列的中国南方和北方双斑长跗萤叶甲地理种群单倍型系统发生树(1 000次重复)
2.4 种群遗传分化与基因流
14个双斑长跗萤叶甲地理种群间总体Fst为0.2481,总体Nm为0.76,说明种群间遗传分化程度普遍较高,种群间基因流弱。从各种群间这两项参数来看,种群间遗传分化指数Fst数值在0.0102~0.8239之间,其中有11对种群间无明显的遗传分化(Fst<0.05),占比12.09%;30对种群间存在中等水平的遗传分化(0.05
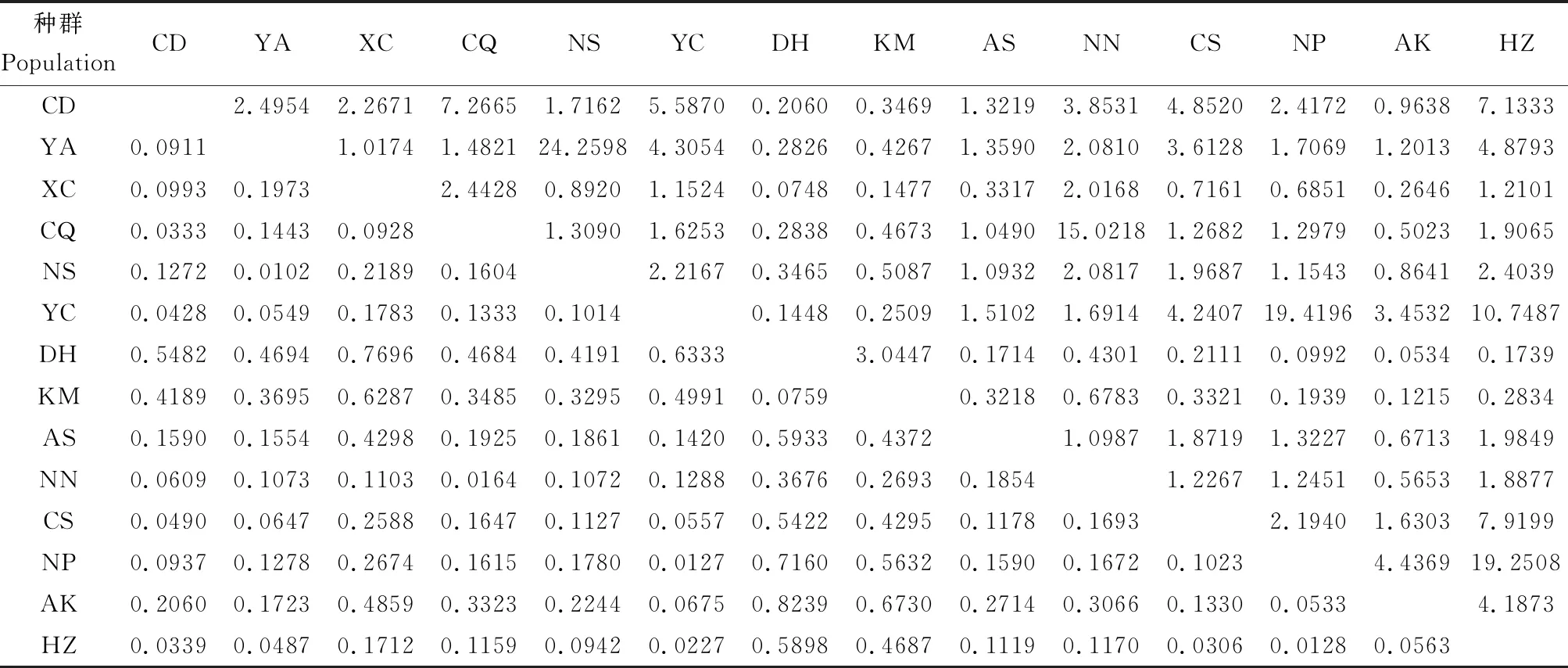
表3 基于COII基因序列的中国南方双斑长跗萤叶甲地理种群间遗传分化指数(Fst)(下三角)与基因流(Nm)(上三角)
AMOVA结果显示COII基因遗传变异种群间方差组分为0.4978,所占方差比率为26.25%;种群内方差组分为1.3986,所占方差比率为73.75%,二者均为极显著水平(表4),表明双斑长跗萤叶甲种群的遗传分化主要来自种群内部个体间的变异,其次才是来自种群间的分化。
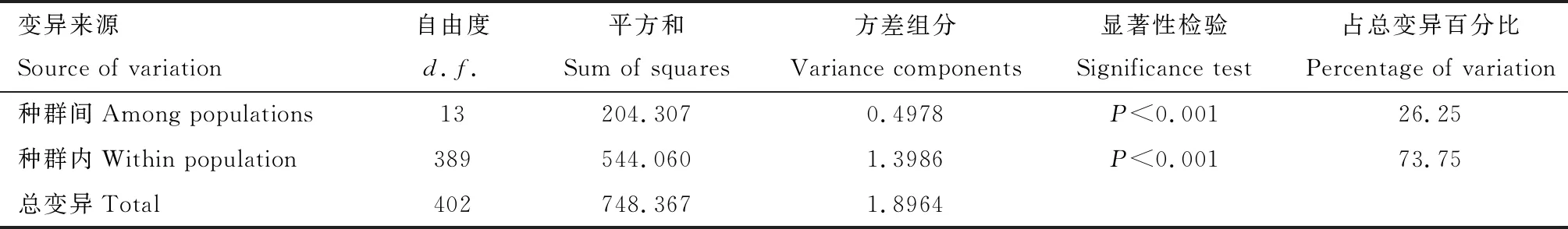
表4 基于COII基因序列的中国南方双斑长跗萤叶甲地理种群变异分子方差分析
为了检测双斑长跗萤叶甲不同地理种群之间的遗传分化程度是否与地理隔离相关,对两两种群间的遗传距离与地理距离(取自然对数)之间的相关性进行Mantel检验,得到遗传距离与地理距离之间的线性回归方程为y=0.103x-0.435,相关系数R为0.2898(P=0.0640>0.05,1 000次随机抽样),表明双斑长跗萤叶甲种群间的遗传分化与地理隔离之间无显著相关性(图3)。
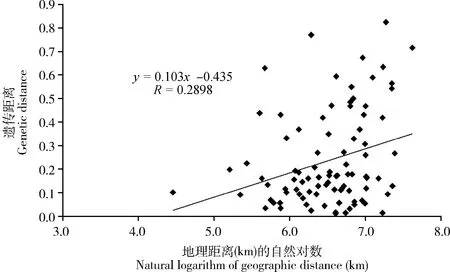
图3 中国南方双斑长跗萤叶甲地理种群间遗传距离与地理距离对数相关性分析
2.5 Wolbachia感染率及感染类型
对14个地理种群403头双斑长跗萤叶甲个体进行Wolbachia共生菌感染检测,结果发现共有396头感染,各种群内感染率在92.59%~100%之间不等,平均感染率为97.60%,其中有7个种群感染率高达100%(表5)。可见Wolbachia在双斑长跗萤叶甲内的感染相当普遍,在种群中均具有很高的感染率。
wsp基因序列分析发现供试双斑长跗萤叶甲种群中共感染了6种Wolbachia株系(strain),根据通用命名法则将其命名为wMhie1~wMhie6,6个株系克隆获得的wsp片段序列长度分别为624, 621, 651, 606, 594和636 bp(GenBank登录号: JF747223-JF747228),wsp基因遗传相似度在78%~93%之间,遗传距离在0.025~0.150之间(表6)。在感染株系中,wMhie1株系在双斑长跗萤叶甲种群中的感染率最高,占检测个体的62.86%;其次是wMhie2,占检测个体的26.43%;其他4种株系在种群中的感染率均较低,wMhie3仅在5个种群中检测到(占检测个体的5.00%),wMhie4和wMhie5仅在3个种群中检测到,而wMhie6仅在2个种群中检测到(表5)。
系统发育分析结果显示,双斑长跗萤叶甲中感染的6种Wolbachia株系(即wMhie1~wMhie6)全部属于A大组,但在系统聚类上与A组中已知的代表性株系分开而单独聚为一个分支,在这个分支内部又表现为wMhie4与wMhie6聚为一支,另外4个株系聚为另一支,且wMhie株系聚类分支结构的置信度分值均高于90,可见此聚类结果具有较高可信度(图4)。基于以上结果,认为双斑长跗萤叶甲中感染的6种株系均属于不同于其他昆虫物种中感染的Wolbachia类群。
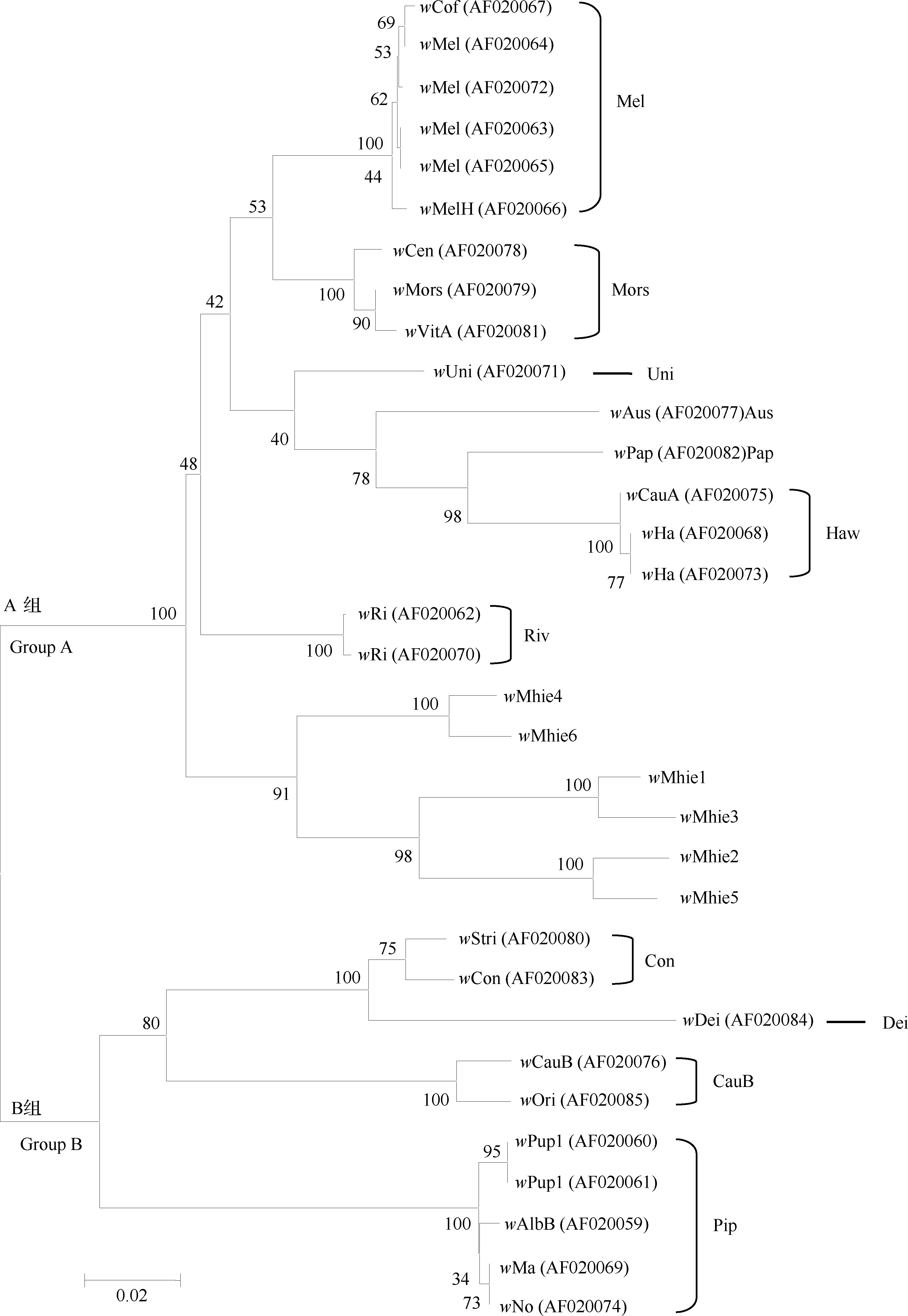
图4 邻接法构建的基于wsp序列的Wolbachia系统发育树(1 000次重复)
3 讨论
本研究以线粒体COII基因作为分子标记,对中国南方多省区分布的双斑长跗萤叶甲地理种群的遗传结构、遗传多样性及种群间遗传分化程度进行了综合分析。在先前报道的北方种群数据结果基础上,又检测到13种新的COII基因单倍型。与梁日霞等(2011)报道的北方种群相比,无论是总群体还是各种群间的平均值,南方种群在单倍型数、单倍型多样性、核苷酸多样度和核苷酸平均差异数等几项参数指标上均明显高于北方种群,说明双斑长跗萤叶甲南方种群的遗传多样性普遍高于北方种群,即南方种群具有更丰富的遗传多样性。造成南北方种群遗传多样性存在差异的原因可能与环境温度相关,已有研究发现双斑长跗萤叶甲的发育历期与温度显著相关,其卵、幼虫和蛹的发育速率随温度升高而加快(李广伟等, 2010; 张聪, 2012)。双斑长跗萤叶甲在我国北方为一年发生1代,以卵在土壤中越冬(张聪等, 2013, 2014),而其在我国南方地区的发生代数迄今尚未有研究报道,推测该种昆虫与鞘翅目其他甲虫相似,即随纬度降低年发生代数增加,因此较发生代数少、发育历期长的北方种群,南方种群内部发生遗传变异的几率增加,种群内具有更丰富的遗传多样性。
遗传分化指数Fst可反映出种群间的遗传分化程度,通常Fst值越大说明群体间遗传分化程度越高。普遍认为当Fst<0.05时群体间遗传分化程度很低,当0.05
对比双斑长跗萤叶甲北方种群的数据,本研究在南方种群中检测到更加丰富的线粒体基因单倍型,从单倍型网络关系图(图1)和系统发生树的结构(图2)来看,MHap14, MHap1和MHap3这3种单倍型在种群中出现频率最高,且均位于单倍型的主干分支中,说明这3种单倍型作为该昆虫种群中的优势单倍型在群体中较稳定的遗传下来。而有7种单倍型构成了一个与主干分支距离较远的丛支,其中的单倍型与多数单倍型之间存在较多的变异位点。这些稀有单倍型的存在丰富了种群的遗传多样性,在一定程度上体现了昆虫种群具备应对环境变化的进化潜力。在主干单倍型与稀有单倍型之间尚存在若干“过渡”单倍型在供试个体中未检测到,可能这些缺失的单倍型(变异位点)由于存在个体稀少已逐渐被淘汰或由于采样量的限制未被检测到。
本研究供试双斑长跗萤叶甲种群中性检验结果大多为负值且不显著(表2),因此不支持种群数量消减或增长、经历瓶颈效应或发生大规模迁移事件的假设,在进化上遵循中性模型,表明种群在较近的历史时期内没有经历明显的群体扩张事件,种群大小保持相对稳定(Korneliussenetal., 2013),此推论与北方种群的检验结果(梁日霞等, 2011)一致。双斑长跗萤叶甲为多食性昆虫,寄主植物种类丰富,因此能应对环境的变化而保持种群的稳定,另外该种昆虫飞行能力弱,在一定程度上制约了种群的迁移和扩张,这也是维持种群相对稳定的另一个原因。
有研究提出Wolbachia在宿主中的感染率常遵循“或多或少(most or few)模式”,即一个物种内的Wolbachia感染率通常会很高(>90%)或很低(<10%)(Hilgenboeckeretal., 2008)。对叶甲科昆虫Wolbachia感染已有一些研究,Ali等(2018)通过检测发现椰心叶甲Brontispalongissima5个东南亚种群中Wolbachia感染率高达100%,且感染类群十分丰富。本研究发现,中国南方双斑长跗萤叶甲所有供试种群中的Wolbachia感染率均高于90%,其中有一半种群100%感染,整体平均感染率高达97.60%(表5),证明Wolbachia在双斑长跗萤叶甲宿主中的感染率符合上述规律。通过wsp基因序列共检测到6种Wolbachia株系,表明在双斑长跗萤叶甲宿主中感染着丰富的Wolbachia类群。系统进化分析显示双斑长跗萤叶甲中感染的6种株系均与目前已知感染昆虫的代表性株系在系统进化上关系较远(图4),尚无法在系统发生上对其准确划分类群,且这6种Wolbachia株系是否具有对双斑长跗萤叶甲宿主具有生殖调控作用仍然未知。
近年来越来越多的研究发现,Wolbachia对其昆虫宿主的调控作用可能在宿主种群发生遗传分化过程中起到了重要的作用,其中一个潜在机制是Wolbachia诱导的胞质不亲和能够有效阻碍感染不同株系的宿主种群之间进行基因交流,从而加速了宿主种群间的遗传分化,甚至可能促进新物种的形成(Jäckeletal., 2013; Zhangetal., 2013; Lisetal., 2015)。本研究发现双斑长跗萤叶甲种群间普遍存在遗传分化且基因交流不频繁,这种现象是否与Wolbachia的生殖调控作用相关仍是未来研究中需要解答的一个问题。在今后的研究中,还需要通过MLST(多位点序列分型)系统对双斑长跗萤叶甲中感染的Wolbachia类群进行更加系统的鉴定,并将Wolbachia感染类群与宿主线粒体单倍型进行结合分析,进一步探究Wolbachia感染是否在双斑长跗萤叶甲种群间产生遗传分化过程中发挥了作用。
猜你喜欢
清华金融评论(2022年4期)2022-04-13
河南农业·综合版(2022年2期)2022-03-18
河南农业(2022年2期)2022-03-14
江西农业学报(2021年8期)2021-09-08
国际放射医学核医学杂志(2021年10期)2021-02-28
上海师范大学学报·自然科学版(2020年5期)2020-12-18
猪业科学(2020年6期)2020-12-11
房地产导刊(2020年7期)2020-08-24
天津农林科技(2020年1期)2020-04-08
植物研究(2020年6期)2020-03-05
