
气相色谱分析方法开发经验总结
2021-07-14 07:07:26长征纵横
实验与分析 2021年2期
文/ 长征纵横
气相分析方法开发的十二个步骤// 方法开发是个复杂而又神秘的工程,往往需要扎实的理论知识储备,但同时也少不了经验的积累;对于方法开发,需要我们从多学科出发,分析仪器学、色谱学、药学、有机化学、分析化学等学科综合,也需要我们熟练掌握文献信息检索,从中吸取精华,同时不断去试错,下面就把我个人职业生涯的经验进行总结提炼,供同行一起学习探讨。
步骤一、确认主要工作内容
1.残留溶剂检测分析方法:GC-HS方法、GC直接进样方法。
2.基因毒杂质检测分析方法。
3.高沸点样品杂质分离与含量检测方法。
步骤二、信息收集
1.要确定待测物质的溶解性、极性、沸点,主要针对“溶剂残留”。
2.需要确定样品的溶解性、热稳定性、极性、沸点、化学性质(酸碱)、物理属性(固体/液体/半固态)。
3.收集并计算化合物(待测物质)的CH含量%比值,CH含量%比值越高检测灵敏度就越好,反之,CH含量%比值越低检测灵敏度就越差。
步骤三、色谱柱使用选择指南(分离关键)
1.非极性色谱柱:分离主要根据沸点高低,沸点低先出,沸点高后出;同沸点的含有O含有N的极性先出,烃类后出,如:DB-1、HP-1、BP-1、SE-30;
2.极性色谱柱:分离主要根据分子极性大小,极性小先出,极性大后出,如DB-Wax;
3.首选中极性毛细管色谱柱,既能兼顾沸点差异又能兼顾极性差异,如:DB-624、HP-5,表1列举了几种常见色谱柱的极性及使用范围;
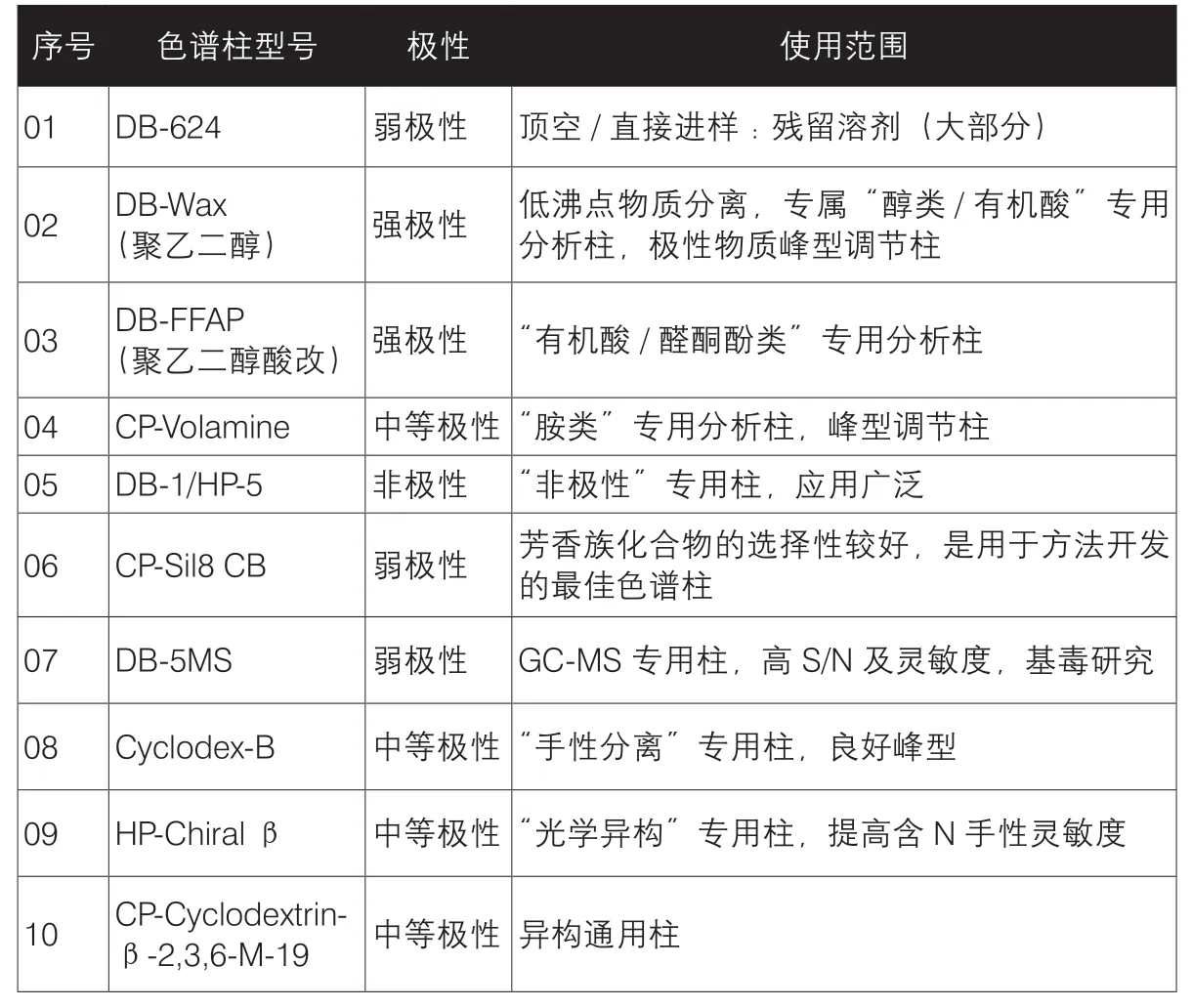
表1 常见色谱柱的极性与使用范围
4.色谱柱直径(内径/粒径)越窄/越小,柱效越高,峰展宽越小,峰越尖锐,峰灵敏度越高,增加分离度;
5.色谱柱膜厚影响传质阻力,液膜越薄的传质阻力越小,在同样的线速度下能够获得更高的柱效,柱效越高,峰展宽越小,峰越尖锐,峰灵敏度越高,增加分离度;液膜厚,保留时间延长。
步骤四、进样方法的选择
1.方法有溶液直接进样法、顶空进样法。
①残留溶剂-HS:大多数是低沸点物质(醇/酯/烷烃/醚),90%以上采用顶空进样(GC-HS),清洁进样,基本不出杂峰(溶剂),原料不被破坏但经济费用高。
②残留溶剂-DS:5%及少数采用溶液直接进样(GC-DS),虽然经济,但出杂峰多(主要原料破坏带入),易损坏色谱柱,需清洁老化保护,表2总结了一些方法开发选择需要考虑的方面。
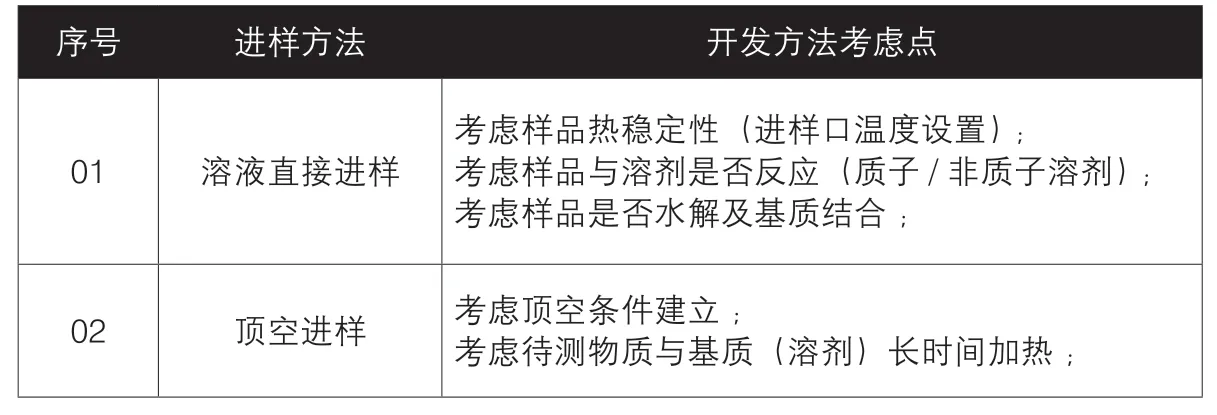
表2 开发选择考虑总结
2.溶剂沸点过高(沸点大于180℃),且样品对热比较稳定,首选溶液直接进样法。
3.溶剂沸点低(沸点小于150℃),且样品对热不稳定,首选顶空进样法。
4.顶空进样法可消除因溶液直接进样会产生降解杂质峰而干扰目标峰(进样口温度大于200℃),采用顶空进样方法还可以减少对样品的预处理。
5.基因毒杂质方法开发,优先选择溶液直接进样法(限度低,主要考虑提高灵敏度);样品杂质分离选择使用溶液直接进样法。
6.对于基因毒杂质限度小于2 ppm以下方法开发,优先选择GC-MS溶液直接进样法。
7.查看CH含量%比值选择进样方法:
①CH含量%比值高:对于残留溶剂检测选择顶空进样法,对于杂质分离/控制或基因毒杂质控制选择溶液直接进样法时,可开发低浓度供试品溶液。
②CH含量%比值低:对于残留溶剂检测选择溶液直接进样法,对于杂质分离/控制或基因毒杂质控制选择溶液直接进样法时,可开发高浓度供试品溶液。
步骤五、确定定量方法
1.外标法:残留溶剂、含量(准确)。
2.内标法:残留溶剂(主要针对易挥发烷烃类)。
3.面积归一化法:样品杂质分离及纯度。
步骤六、样品预处理方法
1.气相色谱仪能直接分析的样品通常是气体或液体,固体样品在分析前应先溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐:进样口温度不能保证盐汽化),往往会聚集在进样口衬管或分流平板导致污染/堵塞,还有可能通过载气进入色谱柱中,损坏色谱柱的填料。
2.样品预处理/溶解原则:根据相似相溶原理。
3.样品分类:气体样品、液体样品、固体样品。
4.气体/液体样品预处理方法:
①直接进样分析(纯度分析与检测),如:甲烷、乙醇;
②脂溶性液体样品(杂质),通常需要用脂溶性有机溶剂溶解样品,如:甲醇、乙醇、乙酸乙酯、二氯甲烷、丙酮、正己烷、
DMSO、DMF;
③进样方式:溶液直接进样、顶空进样。
5.固体样品(含盐)样品预处理方法:
①样品性质:易溶于水,难溶于有机溶剂,样品熔点高,难汽化,如:钠盐/钾盐/盐酸盐/硫酸盐/磷酸盐/富马酸盐;
②如果选择水作溶剂-尽量使用顶空进样方法;
③灵敏度不理想且限度低(1~10 ppm)-可使用水溶解二氯甲烷/正己烷萃取法来进行预处理样品,这样既可以使用溶液直接进样又可以使用顶空进样,检测杂质/残留/基因毒通用。
6.固体样品(不含盐)样品预处理方法:根据相似相溶原理。
步骤七、溶剂系统选择
1.如果是溶液直接进样法,优先考虑溶剂选择,溶解样品的溶剂和最大溶解限度;如果是顶空进样法,可根据样品和溶剂都能溶解;
2.原则上水溶性样品优先选择水,非水溶性样品可选择酯/醇、DMF、DMSO、NMP、DMAC;
3.溶液直接进样-样品溶剂选择考虑:
①化学稳定(水解/醇解),优先选择水(水溶性)/醇/酯(脂溶性)作为溶剂;反之选择烷烃、质子溶剂(万能溶剂:DMF、DMSO、NMP、DMAC)作为溶剂;
②样品具有酸/碱性,但是检测残留溶剂或杂质也具备酸/碱性,可根据待测物质酸/碱性质选择酸碱中和样品;如盐酸/硫酸/富马酸盐等产品检测胺类,需要单独使用碱性溶剂进行中和解离,同样反之,如吡啶/钠/钾盐等产品检测酸类,需要单独使用酸性溶剂进行中和解离,具体溶剂选择需要进行加标回收验证实验。
③供试品溶液要澄清,不与样品及待测物质反应或结合。
4.顶空进样-样品溶剂选择考虑:
①化学稳定(水解/醇解),优先选择水(水溶性)作为溶剂;反之选择质子溶剂(万能溶剂:
DMF、DMSO、NMP、DMAC);
②样品具有酸/碱性,同上“3.②”。
③样品溶解完全没要求,顶空进样气体(溶剂残留)。
5.对于低限度燃烧值大的物质,可选用盐析溶剂系统,如:苯残留、三氯甲烷。
6.样品溶解溶剂选择,也要考虑残留溶剂的溶解度(如:烷、醚类不溶解水)。
7.大部分溶剂系统优先选择DMSO、NMP、DMF,因为既能溶解样品又能溶解溶剂。
步骤八、柱温开发研究
1.等温:对于溶剂个数少(1~3个),且各溶剂沸点差异不算很大,此时可选择等温法,比如50℃保持10~15分钟,适合残留溶剂检测、基因毒杂质检测、含量测定。
2.程序升温:对于溶剂个数多(大于5个),且各溶剂沸点差异较大,此时可选择程序升温法,比如40℃保持10分钟,以10℃/min速率升至180℃,保持5分钟,适用于产品杂质分离、残留溶剂检测,表3、表4展示了经典的程序升温。

表3 经典程序升温(残留溶剂、杂质分离)
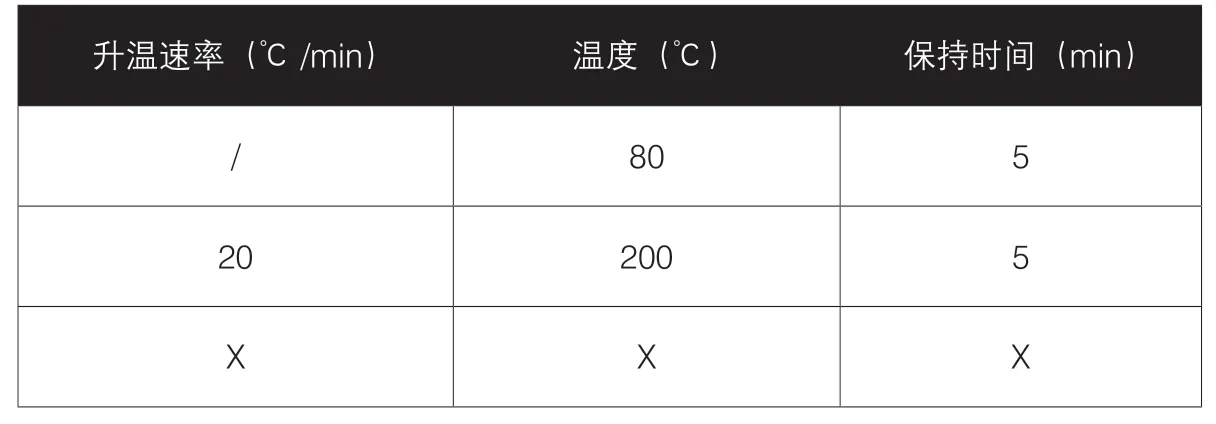
表4 杂质分离的程序升温
3.极端分离:对于大部分沸点低溶剂(当使用DB-624)且考虑分析时长时,可选用低温程序升温法,比如初始温度为35~40℃,分析环境一定要保证温度<25℃。
步骤九、顶空条件方法选择和研究
1.炉温(加热-顶空瓶平衡温度):
①以水为溶剂,温度范围选择75~90℃,最高选择90℃;
②以DMSO、DMF等为溶剂,温度范围选择80~145℃,最高选择140℃;温度过高要考虑高温时长导致溶剂反应降解产生新的未知杂峰;
③考虑温度过高有爆瓶危险,特别在使用盐析方法时(因为溶剂系统里有水分存在)爆瓶概率增加;
④加热温度研究最高温度一般低于溶剂10℃(在考虑灵敏度情况下)。
2.定量环温度:温度要大于炉温10℃。
3.传输线温度:温度要大于定量环温度10℃。
4.顶空瓶(加热)平衡时间:根据大部分溶剂沸点,沸点都相对高平衡时间就长,沸点都相对低平衡时间就短;常规平衡时间为30~60分钟,经典平衡时间为30分钟。
十、检测器选用
1.90%分析方法工作优先选用FID检测器:含碳氢化合物。
2.基因毒杂质选用FID-MS检测器。
3.如含电负性基团(F/Cl/Br)且含CH量少,选用ECD电子捕获检测器,如三氯甲烷无H物质。
4.含有非碳氢化合物组分时,且对检测灵敏度要求不高,通常选择TCD检测器,主要是气体检测,如辅材:氮气。
5.其他:N/P(氮/磷)检测器。
步骤十一、方法建立
1.进样口温度:一般选择200~230 ℃,进样口温度要大于传输线温度,可根据样品性质研究选用中高温,如150~180 ℃(考虑样品不被热解)。
2.检测器温度:一般选择230~260℃。检测器温度要大于进样口和色谱柱温度。
3.流速:3.0~5.0 mL/min,经典流速为:4 mL/min和5 mL/min。
4.分流比:①5:1-50:1;②选择规则:响应值高,使用大分流比,反之,则使用小分流比;③经典分流比为5:1(顶空进样)和50:1(溶液直接进样)。
5.进样量:顶空进样为1 mL,溶液直接进样为0.2-1 uL,也可根据进样峰型进行改进。
6.样品浓度/配制开发研究:
①顶空进样:0.2~1 g(根据限度/灵敏度要求进行研究),对于大浓度要考察样品提取平衡;浓度一般控制在0.1~0.4 g/mL(顶空装量控制在1~5 mL);
②溶液直接进样:一定要考虑溶解完全,浓度可根据限度/灵敏度要求进行开发研究;一般控制在0.5~200 mg/mL;
③开发研究合适的样品浓度,一定要根据杂质限度/灵敏度做相应研究调整。
7.对于杂质分离(溶液直接进样法),在方法开发同时也要考察残留问题,可增加清洗老化色谱柱方法程序,也可增加清洗进样针方法;还有对于对气相管路有吸附残留性质也可增加清洗程序(如胺类、>150 ℃高沸点物质),增加程序一定要作为分析方法的一部分,且一定要经过验证验收。
8.方法开发时遇到难点:分离度、灵敏度、峰型。
分离度优化研究:
①极性色谱柱与非极性色谱柱的转换使用(DB-Wax与DB-624转换);
②使用程序升温(速率可低至3~5 ℃/min,但是分析时长增加,灵敏度减小);
③使用长色谱柱(如30 m→45 m→75 m);
④使用小粒径色谱柱(3 um→1.8 um→1 um);
⑤降低流速(变流速如:3→1 mL/min);
⑥使用程序降温(120→40℃,速率10-20 ℃/min)。
灵敏度优化研究:
①增加进样量(1→2uL);
②增大流速;
③增加顶空瓶装样量(最大5 mL,可研究6~8 mL);
④使用小粒径色谱柱(3 um→1.8 um→1 um);
⑤使用盐析方法(成功典型:苯、三氯甲烷);
⑥顶空方法:适当增加顶空瓶加热温度和时长(进行对比研究)。
峰型优化研究:
①使用小粒径色谱柱;
②增大柱流量;
③使用专属柱(如胺类分析柱、醇类分析柱);
④升高柱温。
9.系统干扰考察研究:先走空白2针:样品1针、对照1针,查看空白、样品溶液的干扰情况,空白与样品应无干扰峰出现(方法验证-专属性),且对LOQ和LOD测试无干扰,基本能通过LOQ和LOD验证(方法好坏体现在系统与溶剂对测试的干扰)。
10.系统有干扰可通过换溶剂或调整程序升温来消除对目标峰的干扰。
11.进行LOQ考察:只确认灵敏度(响应值)最小的2个,将LOQ试验出来,LOQ对应浓度是否合格(LOQ含量不得高于限度的50%)。
12.最后进行100%加标回收测试,回收结果如果满足90%~105%,方法基本确定。
步骤十二、分析方法评价与挑战
1.分析方法开发评价标准:
①空白考察:空白溶液无干扰峰出现,应该对测试无影响(最小干扰<LOD);
②分离度:最低分离度标准为,各相邻峰间分离度>2.0(最小1.5);
③LOQ:确认灵敏度(响应值)最小的2个,将LOQ试验出来,LOQ对应浓度是否合格(LOQ含量不得高于限度的50%),且LOQ的S/N要大于10;
④加标回收标准(90%~110%);
⑤溶液稳定性:100%加标溶液考察0、2、4、8、24小时的稳定性,主要考察峰面积RSD%(n=5)以及各溶剂的回收率%。
2.分析方法简单预实验(预验证):
①线性:50%、100%、150%、200%(每点进2针),R大于0.999;
②准 确 度:50%、100%、200%(每点进2针),回收率%应在90%~110%;
③SST实验:6针对照结果,各溶剂峰峰面积RSD%(n=6)均小于6.0%,且考察各溶剂峰的拖尾因子或对称因子(0.9~1.1,最大不可超过2.0)。
猜你喜欢
艺术品鉴(2020年6期)2020-12-06 10:49:08
中国经济周刊(2020年24期)2020-01-15 14:31:06
分析化学(2018年12期)2018-01-22 12:31:46
中学生数理化·高二版(2017年3期)2017-07-07 21:55:13
领导文萃(2017年6期)2017-03-24 09:31:39
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
食品工业科技(2014年9期)2014-03-11 18:15:44
中国烟草学报(2012年3期)2012-04-10 12:50:46
中国烟草学报(2012年6期)2012-04-09 07:41:40
