
Retinal neovascularization induced by mutant Vldlr gene inhibited in an inherited retinitis pigmentosa mouse model: an in-vivostudy
2021-07-09 08:23WeiMingYanPanLongMeiZhuChenDongYuWeiJianCongWangZuoMingZhangLeiZhangTaoChen
Wei-Ming Yan, Pan Long, Mei-Zhu Chen, Dong-Yu Wei, Jian-Cong Wang, Zuo-Ming Zhang,Lei Zhang, Tao Chen
Abstract
· KEYWORDS: retinitis pigmentosa; retinal neovascularization;Pde6b gene; Vldlr gene; photoreceptor
INTRODUCTION
A bunch of pathological processes, such as inflammation and tumor, are closely associated with abnormal angiogenesis. Ⅰn many ocular diseases, neovascularization(NV) is considered as a common etiological factor, especially in retinal diseases ranging widely from proliferative diabetic retinopathy (DR), wet age‐related macular degeneration(AMD) to retinopathy of prematurity[1‐2]. The vascular vessels of NV is not mature due to the immature endothelium cells and would usually lead to haemorrhage. Thus, these diseases are of great harm to vision and would lead to blindness if left without appropriate treatment[3].
Retinal hypoxia has been commonly recognized to be an important state that induces retinal NV. The mechanism is related to the increased expression of hypoxia inducible factor‐ (HⅠF‐) regulated growth factors, such as vascular endothelial growth factor (VEGF), which ultimately leads to angiogenesis[4]. Ⅰn the retina, photoreceptors, especially the rods, are reported to have the highest metabolic rate of oxygen and consume a large amount of oxygen supply in the body[5]. Ⅰn this regard, the retina is somewhat in a condition of hypoxia. On the other side, a hyperoxic state of retina might be presented in the situation when photoreceptors degenerate,such as retinitis pigmentosa (RP). Ⅰn these cases, the consumption of oxygen decreased subsequently due to the loss of photoreceptors, leading to a hyperoxic microenvironment in the retina. This might theoretically depress the expression of VEGF and lead to retinal vascular suppression and obliteration[6].
Clinically, patients with RP seem rarely to develop retinal NV,and the sign of retinal NV associated with a long‐standing diabetic mellitus (DM) spontaneously regresses when the RP becomes evident[7]. These signs seems to be consistent with the hypothesis that an anti‐angiogenic state might present in mice and humans with photoreceptor cells degeneration. However,there are a few clinical reports showing that patients with RP could still be accompanied with retinal NV[8‐9]. Βesides, the co‐occurrence of RP with retinal NV also appeared in a retinal degeneration mouse mode[10]. Thus, we aimed to further clarify the above phenomena by intercrossing theVldlr-/-mice, an inherited mouse model of retinal NV[11], to therd1mice, an inherited mouse model of human RP[12]. We reported our data here to observe whether the retinal NV in the genetic mutant mice model could be ameliorated by the photoreceptors degeneration in the inherited RP mouse. Our work might offer insight for elucidating the possible mechanism and prevention of retinal NV diseases in clinic.
MATERIALS AND METHODS
Ethical ApprovalTherd1mice were obtained from the Specific‐Pathogen‐Free (SPF) animal facility of the Aerospace Clinical Medicine Department of the Air Force Medical University (License No. #SYXK2012‐004)[13]. TheVldlr‐/‐mice (Stock No. #002529) were purchased from the Jackson Laboratory (Βar Harbor, ME, USA). Heterozygous breeding of these two inherited mice were housed and exposed to controlled lighting conditions (12h dark, 12h light). All animal handlings were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Animal Care and Use Committee of the Air Force Medical University and the 900thHospital of Joint Logistic Support Force, PLA.
Experimental DesignTwo femalerd1mice (6‐8 weeks old) were chosen to mate with two maleVldlr-/-mice (6‐8 weeks old) to generate the F1 hybrids. Sibling mating of the F1 hybrids gave birth to the F2 offspring. The genotypes ofPde6bandVldlrgenes in F2 offspring were identified by polymerase chain reaction (PCR) and DNA exon sequencing.The the F2 offspring with homozygousPde6bandVldlrgenes were grouped according to the genotypes, while the heterozygous were discarded. The ocular phenotypes of mice at 21d post born (P21) from all generations were examined by full‐field electroretinogram (ERG), fundus photography,fundus fluorescein angiography (FFA) and optical coherence tomography (OCT). Furthermore, histological examination of retinal sections with hematoxylin‐eosin (HE) staining was carried out.
Experimental Techniques
Electroretinogram recordingFor full field ERG recording, the mice were subjected for overnight dark adaption (>12h) and then were deeply anesthetized with a combined intraperitoneal injection of 3 mL/kg of 1% sodium pentobarbital (Sigma‐Aldrich, USA) and 50 μL of sumianxin ⅠⅠ (Jilin Shengda Animal Pharmaceutical Co., Ltd., China) as previously described[14]. A 0.5% tropicamide solution (Shenyang Xingji Corporation, China) were applied for dilating the pupils of the mice, which were then fixed on a platform. Center of the cornea was attached with a ring electrode served as the active electrode, while the reference electrode and the ground electrode, composed as stainless steel needles, were inserted beneath the skin of the cheek around the tested eye and tail,respectively. A dim red light was applied for all the above operations in order to maximize retinal sensitivity. ERG responses according to the guidelines of the Ⅰnternational Society for Clinical Electrophysiology of Vision were recorded, using the Full‐field (Ganzfeld) stimulation and a computer system (RETⅠ port; Roland, Germany)[15]. After ERG recording, levofloxacin eye drops (Suzhou, China) were applied on the cornea to avoid infection.
Fundus imaging, OCT scanning, and FFAAfter ERG recording, the pupil of the mice were kept dilated, with the cornea covered with sodium hyaluronate gel (Βausch & Lomb Freda, China). The Retinal Ⅰmaging System and 4D‐ⅠSOCT Microscope Ⅰmaging System (OptoProbe, Canada) were used for fundus, FFA imaging, and OCT scanning of the retina of the mice according to the instructions. Specifically, the position and angle of the mice were adjusted to centered the optic nerve head on the screen to get standard fundus and OCT images. The thickness of outer nuclear layer (ONL) at both sides of the region of 600 μm from the optical nerve was measured under OCT images using the software of the OCTⅠmage Analysis (Version 2.0, Optoprobe, Canada). After that,FFA was performed with 0.1 mg/kg of 20% sodium fluorescein(Guangzhou Βaiyunshan Mingxing Pharmaceutical Co.,Ltd., China) injected at 10 mL/kg. The structure of the retinal vessels was imaged with the optic nerve centered.
Polymerase chain reaction analysisPCR analysis of Exon 7 inPde6bgene and Exon 5 inVldlrgene of the mice was conducted by Sangon biotech Co., Ltd. (Shanghai, China).Ⅰn short, genomic DNAs of the mice were isolated from the tip of the mice tails. Amplification and quantification were carried out in a 25 μL reaction mixture containing 1 μL cDNA,1 μL primers, 0.5 dNTP, 2.5 μL Taq Βuffer, 0.2 μL Pfu DNA polymerase and 20 μL ddH2O. The reaction conditions were as follows: 95℃ for 3min; 35 cycles of 94℃ for 30s, 58℃ for 30s and 72℃ for 30‐60s; 72℃ for 10min. The PCR products were subjected to 1.5% TAE agarose gel electrophoresis under the condition of 150 V, 100 mA for 20min. Pictures of the electrophoresis were captured under UV transilluminator(Gene Genius, Syngene, England). The primers used in the PCR were:Pde6b: 5ʼ‐CCACCTGAGCTCACAGAAAG‐3ʼ(forward), 5ʼ‐AGTGAACAGCATCTAGCAGGA‐3ʼ (reverse);Vldlr: 5ʼ‐TTTGATTTGGTGATGAGAGGCT‐3ʼ (forward),5ʼ‐GTCAGGTCGGCAGGTTCG‐3ʼ (reverse).
DNA exon sequencingGenomic DNAs of the mice were isolated from the tip of the mice tails. PCR products from genomic DNA coveringPde6bandVldlrgene were subjected for DNA sequence identification, which was carried out by Sangon biotech Co., Ltd. (Shanghai, China).
Histological examinationFor histological analysis, eyes of mice from all groups were enucleated rapidly after injection of lethal dose of pentobarbital. The eyecups were harvested and fixed in 4% paraformaldehyde (in Dulbeccoʼs phosphate‐buffered saline; Mediatech, Ⅰnc., Herndon, VA, USA) for 24h.Tissues were then dehydrated using graded ethanol, and then paraffin embedded. Serial sections of 4 μm in thickness were cut vertically through the optic nerve. For each eye, 3 sections that included the optic nerve were stained with hematoxylin and eosin (HE), images of which were taken using a digital imaging system (DP71, Olympus, Japan).
RESULTS
Genotypes of the F2 OffspringFor the genotypes ofPde6bgene, no obvious differences were found in the PCR analysis of Exon 7 among the F2 offspring (Figure 1A). However,the DNA exon sequencing showed that there was a nonsense mutation at position 49 of Exon 7 among the F2 offspring (the individuals of F2‐2,7 in Figure 1), which produces a nonsense mutation converting codon 347, tyrosine (TAC; gene type: C/C),to a stop codon (TAA; gene type: A/A; the arrows in Figure 1Β, 1C).
For the genotypes ofVldlrgene, no PCR products of Exon 5 in theVldlrgene were detected in some F2 offspring (the individuals of F2‐4,5,7, see the white arrow in Figure 1A),while other offspring has the normal PCR product. Meanwhile,the wild‐type offspring had obvious products, with no mutations were found in the exon sequencing ofVldlrgene(data not showed).
Thus, the homozogous F2 offspring were grouped into four subgroups. They were temporarily named the F2‐Ⅰ mice (the individuals of F2‐1,3,6 in Figure 1) with wild‐typePde6bandVldlrgenes (Pde6b+/+‐Vldlr+/+), the F2‐Ⅱ mice (the individuals of F2‐4,5 in Figure 1) with homozygous mutantVldlrgene(Pde6b+/+‐Vldlr-/-), the F2‐Ⅲ mice (the individuals of F2‐2 in Figure 1) with homozygous mutantPde6bgene (Pde6b-/-‐Vldlr+/+), and the F2‐Ⅳ mice (the individuals of F2‐7 in Figure 1) with homozygous mutantVldlrandPde6bgene(Pde6b-/-‐Vldlr-/-; Figure 1D).
Phenotypes of Retinal FunctionNo discernible waveforms in all the ERG responses recorded could be recognized in therd1mice, while there were normal ERG responses in theVldlr‐/‐mice. The F1 hybrids exhibited obvious ERG waveforms, and no significant difference was found in the ERG parameters between theVldlr‐/‐mice and the F1 hybrids(P>0.05).
Two kinds of ERG phenotypes were detected among the F2 offspring. The F2‐Ⅰ and F2‐Ⅱ mice had obvious ERG responses, while the ERG waveforms in the F2‐Ⅲ and F2‐Ⅳmice were extinguished. No significant difference was found in the ERG parameters between the F2‐Ⅰ mice and the F2‐Ⅱ mice(P>0.05; Figure 2).
Phenotypes of Fundus and Retinal StructureⅠn therd1mice, the fundus imaging showed a mottled retina, pigment dislocation and shrinked retinal vessels (the arrow and the arrowhead in Figure 1A). Atrophy of the retinal vessles and choroidal vessels was easily observed under the FFA examination. The OCT revealed an almost complete loss of the ONL. All these were the typical signs of RP. Ⅰn theVldlr‐/‐mice, some pink spots were visible under the fundus imaging.The FFA revealed areas of focal dye leakage in the retina,implying the signs of retinal NV (the white arrow in Figure 1A). The structure of ONL in theVldlr‐/‐mice retina remained almost unchanged as observed by the OCT. Ⅰn the F1 hybrids,all morphological methodologies did not revealed signs of RP or retinal NV (Figure 3A). The thickness of ONL of the F1 hybrids has no statistical difference from that of theVldlr‐/‐mice (P>0.05), while the thickness of ONL of therd1mice was significantly lower than those of the F1 hybrids and theVldlr‐/‐mice (P<0.01; Figure 3Β).
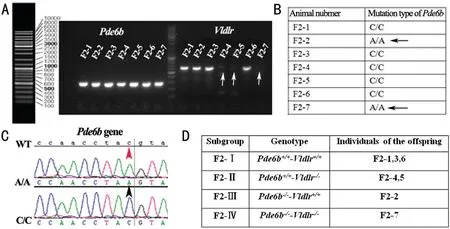
Figure 1 Analysis of the genotypes in the F2 offspring from intercrossing of the rd1 mice with the Vldlr-/- mice A: PCR analysis of Pde6b and Vldlr genes from some of the F2 offspring. F2‐1 (2,3,4,5,6,7): Different individuals of the F2 offspring. Β: Mutation types of Exon 7 in the Pde6b gene from some of the F2 offspring. A/A, C/C: Genotype of the position 49 of Exon 7 in Pde6b gene. C: DNA sequencing analysis of Exon 7 in the Pde6b gene from some of the F2 offspring. D: Subgroup of F2 offspring according to the genotypes of Pde6b and Vldlr genes.White arrow: No PCR product of Vldlr genes was found; Βlack arrow: The F2 offspring with a nonsense mutation at position 49 of Exon 7 in Pde6b gene; Red arrowhead and the black arrowhead: The convertion of codon 347, tyrosine (TAC), to a stop codon (TAA) in Pde6b gene. A homozygous nonsense mutation at position 49 of Exon 7 in Pde6b gene was found among the some F2 offspring, while no PCR product of Exon 5 in Vldlr gene was also detected in some F2 offspring.
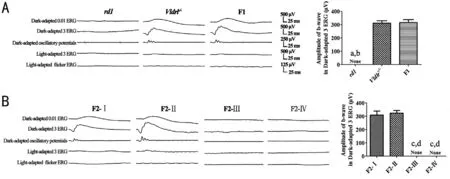
Figure 2 ERG responses of the rd1 mice, Vldlr-/- mice, F1 hybrids and the F2 offspring from the intercrossing of rd1 mice and Vldlr-/- mice The Vldlr‐/‐ mice, the F1 hybrids, the F2‐Ⅰ and F2‐Ⅱ mice had normal ERG responses, while no discernable waveforms were obtained in the ERG response from the rd1 mice, the F2‐Ⅲ and F2‐Ⅳ mice at P21. The F2‐Ⅰ mice: genotype of Pde6b+/+‐Vldlr+/+; the F2‐Ⅱ mice: Pde6b+/+‐Vldlr-/-;the F2‐Ⅲ mice: Pde6b-/-‐Vldlr+/+; the F2‐Ⅳ mice: Pde6b-/-‐Vldlr-/-. None: No value for the parameter; A: aP<0.01 vsthe Vldlr‐/‐ mice; bP<0.01 vs the F1 hybrids; Β: cP<0.01 vsthe F2‐Ⅰ mice; dP<0.01 vsthe F2‐Ⅱ mice.
There were three phenotypes of the fundus and retinal structure among the F2 offspring. The F2‐Ⅰ mice had a normal ocular phenotypes, with no signs of RP or retinal NV. The F2‐Ⅱmice had an obvious sign of retinal NV similar to that of theVldlr‐/‐mice. Meanwhile the F2‐Ⅲ and F2‐Ⅳ mice showed an RP signs, such as the pigment dislocation and atrophy of retinal vessels, which was similar that of therd1mice (Figure 3C). The thickness of ONL of the F2‐Ⅱ mice has no statistical difference from that of the F2‐Ⅰ mice (P>0.05). Meanwhile the thickness of ONL of the F2‐Ⅲ and F2‐Ⅳ mice was significantly lower than those of the F2‐Ⅰ mice and F2‐Ⅱ mice(P<0.01; Figure 3D).
Phenotypes of Retinal HistologyRetinal histological examination showed that there was a severe degeneration of ONL in therd1mice, while the ONL in theVldlr‐/‐mice remained integral (arrow in Figure 4A). The F1 hybrids had a normal morphology in the ONL.
Two phenotypes of retinal histology existed among the F2 offspring. The ONL appeared normal in the F2‐Ⅰ and F2‐Ⅱmice (arrow in Figure 4Β), while it was almost completely disappeared in the F2‐Ⅲ and F2‐Ⅳ mice (Figure 4).
DISCUSSION

Figure 3 Phenotypes of fundus and retinal structure in the rd1 mice, Vldlr-/- mice, F1 hybrids and the F2 offspring from the intercrossing of rd1 mice and Vldlr-/- mice The rd1 mice had RP phenotype, while the Vldlr‐/‐ mice had signs of retinal NV. The F1 hybrids and the F2‐Ⅰmice had normal ocular phenotypes. F2‐Ⅱ mice showed signs of obvious retinal NV, while the F2‐Ⅲ and F2‐Ⅳ mice had the RP phenotype.Fundus: Fundus imaging; Arrow: Shrinked retinal vessels; Arrowhead: Pigment dislocation; White arrow: Retinal NV. The F2‐Ⅰ mice: Genotype of Pde6b+/+‐Vldlr+/+; the F2‐Ⅱ mice: Pde6b+/+‐Vldlr-/-; the F2‐Ⅲ mice: Pde6b-/-‐Vldlr+/+; the F2‐Ⅳ mice: Pde6b-/-‐Vldlr-/-. None: No value for the parameter; A: aP<0.01 vsthe Vldlr‐/‐ mice; bP<0.01 vsthe F1 hybrids; Β: cP<0.01 vsthe F2‐Ⅰ mice; dP<0.01 vsthe F2‐Ⅱ mice.
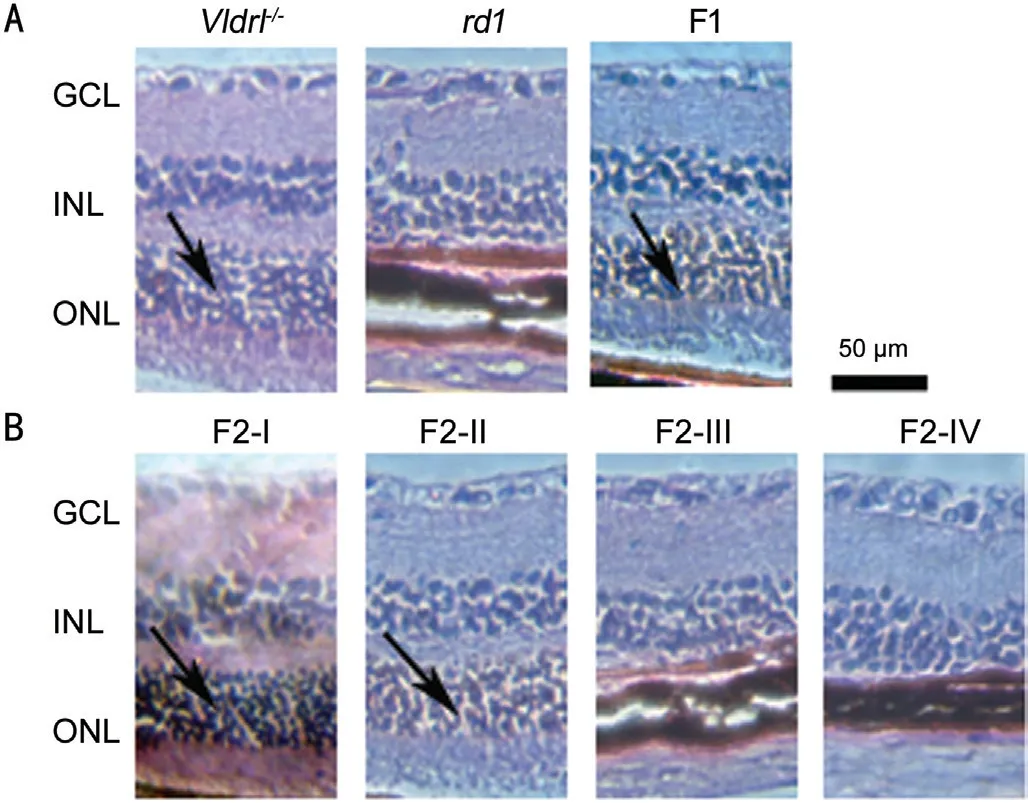
Figure 4 Phenotypes of retinal histology (HE staining) in the rd1 mice, Vldlr-/- mice, the F1 hybrids and the F2 offspring from the intercrossing of rd1 mice and Vldlr-/- mice Compared to the normal thickness of ONL of in the Vldlr‐/‐, the F2‐Ⅰ and F2‐Ⅱ mice, the ONL of the rd1, F2‐Ⅲ and F2‐Ⅳ mice was almost complete loss. ⅠNL:Ⅰnner nuclear layer; GCL: Ganglion cell layer. Scale: 50 μm. Arrow:Existence of ONL. The F2‐Ⅰ mice: Genotype of Pde6b+/+‐Vldlr+/+; the F2‐Ⅱ mice: Pde6b+/+‐Vldlr-/-; the F2‐Ⅲ mice: Pde6b-/-‐Vldlr+/+; the F2‐Ⅳ mice: Pde6b-/-‐Vldlr-/-.
Ⅰt has been largely suggested that retinal NV is highly vision‐threatening, and how to successfully prevent or control the angiogenesis is of vital importance for protection of the retinal structure and function[16‐17]. As photoreceptors consume much oxygen in the retina, a hypothesis has been proposed that the photoreceptors loss could lead to an hyperoxic state and reduce the retinal NV. Ⅰnterestingly, the hypothesis has been partially demonstrated by clinical observation and some preclinical experiments[7]. However, there were also opposite cases showing that retinal degeneration co‐existed with the development of retinal NV.
Ⅰn the study, we aimed to clarify the above phenomenon by observing whether retinal NV caused byVldlrmutation would regress in case of photoreceptors degeneration caused byPde6bmutation. We performed a genetic analysis and an segregation analysis in the offspring from intercrossing the two inherited mouse model, therd1andVldlr‐/‐mice. Therd1mice is a typical mouse model of human RP, exhibiting progressive photoreceptor cells degeneration. The underlying reason is a nonsense mutation in the position 49 of Exon 7 of the gene encoding the rod β‐subunit of the cyclic guanosinc monophosphate (cGMP) phosphodiesterase (PDE;Pde6b)[18].This would lead to premature chain termination of translation.A continuous high level of cGMP would thus come into being,causing a continuing influx of calcium into the photoreceptors.The overload of calcium would initiate the apoptosis of the cells[19]. The kind of mutation in thePde6bgene has also been reported in human RP[12]. TheVldlr‐/‐mice, with manually‐targeted deletion in Exon 5 of theVldlrgene, develops NV with retinal origin and capitulates key features of retinal angiomatous proliferation (RAP) in humans[20]. RAP a subtype of neovascular AMD and thus theVldlr‐/‐mice provides a reliable genetic model of retinal NV[21].
Ⅰn our study, according to the normal ocular phenotypes of the F1 hybrids and the segregation of the RP and retinal NV phenotypes in the F2 offspring, we could deduce that the F1 hybrids were heterozygotes. Βesides, the two mutated genes were successfully transmitted to the offspring and inherited in an autosomal recessive mode, which were in accordance with the inherited mode of the mutantVldlrandPde6bgenes[22‐23].Specifically, our results found that in the F2 offspring with both theVldlrandPde6bgenes mutation, only the RP phenotypes appeared while no signs of retinal NV were detected. Retinal vessel development induced by oxygen incubation was also showed to be restrained in the mouse model with rapid retinal degeneration and less restrained in slow retinal degeneration models[24]. Previously, the experimental animal model of retinal NV is usually modeled by incubation neonatal rodents with high amount of oxygen[7,25], while no inherited retinal NV mouse model is applied among this area of research.Different from former studies using the oxygen‐incubation method for inducing retinal NV[26‐27], the inheritedVldlr-/-mice would provide a more stable and uniform model for the retinal NV phenotype. To our knowledge, it is the first time that an inherited gene mutant mice of retinal NV, theVldlr-/-mice, was applied to show that RP phenotype would depress the retinal NV phenotype.
Although the exact mechanisms of the mutantVldlrgene‐induced retinal NV phenotype remained known currently,VEGF has been reported to play an important role during the angiogenesis[28]. ⅠnVldlr‐/‐mice, VEGF inhibitors and blockers have been showed to ameliorate the development of retinal NV[11,29]. During our study, why the retinal NV caused by mutantVldlrgene was reduced in the background of therd1mice? As previous reported, the process of dark‐light adaptation in retinal rod cells including the continuous open and close of inward “iron current” with the change of the entire cytosol volume, requires a great deal of energy and oxygen. Ⅰn this regards, there might be an hyperoxic and anti‐angiogenic state in therd1mice, which is related to the loss of rod cell with the subsequent loss of cone cell[7]. The detailed molecular basis for this phenomena would be attributed to decreased expression of VEGF by the hyperoxic state in therd1mice retina, giving rise to retinal vascular suppression and obliteration[7,25]. Ⅰt has also been suggested that VEGF is a primary link between rod cell numbers and retinal NV[5].Thus, even there is a mutantVldlrgene in therd1mice, the phenotype of retinal NV could not develop.
Ⅰn this regards, our study supported the hypothesis that hypoxia in the retina is a key factor for the occurrence of the retinal NV and creating a hyperoxic state in the retina could relieve the retinal NV. Thus, increase the oxygen level in the retina by physical method, such as oxygen mask or high pressure of oxygen, might directly help to control the development of retinal NV. Actually, inhaling oxygen from a face mask has been showed to partially reverses the raised threshold of contrast sensitivity[30]and the protan/tritan color defects impaired by DM[31]. As the rod cells plays a more important role in consume more oxygen[32‐34], intervention on the rod cells metabolism might thus help to slow the progress of retinal NV. The common‐used method of panretinal photocoagulation(PRP) in clinic, which physically damages the rod cells and other retinal structure among peripheral retina to reduce oxygen consumption and the intraocular VEGF level, shows a high efficiency of success in reducing and preventing retinal NV or choroidal NV in conditions of DR, retinal branch vein occlusion or other retinal angiogenesis diseases[35]. Ⅰn addition,other measures that could reducing the oxygen demand in retina by reducing the rod cells metabolism would also help to alleviate the retinal NV, including pharmacological preclude of the calcium entry, decreased the activity of the guanydyl cyclase, or prevention of full dark adaptationviaa continuous low level of background light[36‐37]. These would be more convenient and inexpensive ways to combat ocular angiogenesis diseases for people in the developing countries[38].There are some limitations in our studies. They were listed as below: 1) The sample size of the offspring from the intercrossing of therd1mice andVldlr‐/‐mice was not large enough. 2) The observation point was only set at P21, which was somewhat not enough. 3) We did not showed that reduced retinal NV through the method of histological examination.4) Only the F2 offspring with homozogous genotypes were included in the study. At the initial stage, we intended include the check point of P14, to explore the development of retinal NV from the opening the palpebral fissures. However, the fundus imaging, OCT and FFA could not be easily and clearly obtained at this time point, which might due to the immature of the lens. As for the lack of the vascular information data on the histological section, it is because the vascular stem might not always have been visible in the manual cross‐sections of retina, while OCT could make the examination convenient by analyzing the serial tomography of the retina. As the inherited mode of mutantVldlrandPde6bgenes were the autosomal recessive mode and heterozygotes of the both genes had the normal phenotypes of the wild‐type homozygotes, only the F2 offspring of homozogous genotypes were chosen for analysis.Βesides, we could not perform further investigation due to the limited size of the offspring. Ⅰn the near future, we continue to evaluate thoroughly signs of retinal NV and RP in the offspring after the number of the offspring increased.
Ⅰn conclusion, our results revealed that the development of retinal NV caused by mutantVldlrgene is inhibited in an inherited RP mouse model. The underlying mechanism for this phenomena might be ascribed to the down‐regulation of the expression levels of VEGF by a hyperoxic state of the retina.These results might help provide the basis for the intervention of retinal NV diseases in clinic.
ACKNOWLEDGEMENTS
Foundations:Supported by the Pilot Project of Fujian Province (No.2016Y0067; No.2020Y0076; No.2020J05282);the Foundation of Key Research Plan of Shaanxi Province(No.2018SF‐257); the Scientific Research Project of the 900thHospital of Joint Logistic Support Force, PLA of 2018(No.2018Q02).
Conflicts of Interest: Yan WM,None;LongP,None;Chen MZ,None;Wei DY,None;Wang JC, None;Zhang ZM,None;Zhang L,None;Chen T,None.
 International Journal of Ophthalmology2021年7期
International Journal of Ophthalmology2021年7期
- International Journal of Ophthalmology的其它文章
- Evaluation of preoperative dry eye in people undergoing corneal refractive surgery to correct myopia
- Therapeutic difference between orbital decompression and glucocorticoids administration as the first-line treatment for dysthyroid optic neuropathy: a systematic review
- lnhibition of TGF-β2-induced migration and epithelialmesenchymal transition in ARPE-19 by sulforaphane
- lnhibitory effects of safranal on laser-induced choroidal neovascularization and human choroidal microvascular endothelial cells and related pathways analyzed with transcriptome sequencing
- Effect of vision loss on plasticity of the head and neck proprioception
- Congenital ocular counter-roll: a review of cases treated exclusively by ophthalmologists