
基于转录组测序技术的椎间盘退变特异基因表达谱分析
2021-07-08 02:48夏博文邢军超艾秋池李瀚卿徐美涛侯天勇
南方医科大学学报 2021年6期
夏博文,邢军超,艾秋池,李瀚卿,徐美涛,侯天勇
陆军军医大学第一附属医院骨科,重庆 400038
作为骨科常见病,下腰痛是威胁当代人类身心健康的主要因素之一,椎间盘退行性变(IDD)则与很多下腰痛病例具有较高的相关性[1]。椎间盘是椎体间的连接结构,其由位于中央的髓核(NP)、位于边缘的纤维环(AF)以及与上下椎体毗邻的软骨终板(EP)构成,它的主要功能是缓冲人自身体重及运动所产生的机械负荷[2]。然而,在退变状态下,椎间盘将无法很好地满足正常的生理需要[1]。现阶段,保守和手术方法均可用于IDD的临床治疗,前者包括对症的非甾体抗炎药(NSAIDs)、物理疗法和行为调整等,后者则主要是椎间盘摘除及椎体融合术[3-5],但手术病人将承受数月的受限腰部活动和可能发生的邻椎病等并发症[6]。因此,为了改进临床治疗方法和改善病人预后,寻找IDD的关键影响因素并发展对因治疗方法便成了此领域的主要研究方向。
转录组测序(RNA-seq)技术可用来获取某物种细胞或组织的完整转录组数据,从而揭示该物种在不同生长环境下的基因表达变化[7]。既往已有学者使用此方法分析IDD进展过程中的差异基因和通路,但由于临床正常椎间盘样本难以获取,他们将椎间盘突出患者样本和椎体滑脱(患者样本进行对比,具有一定误差[8]。同时,动物模型也被应用于相似的研究中,然而由于人与动物间不同的分子表型和椎间盘结构,其结论仍有争议[9]。
本课题组通过RNA-seq技术,首先对临床手术采集到的椎间盘样本进行测序,其次根据其有无IDD征象分为2组,对比所得的测序数据,得到差异基因,并根据它们在通常生理过程中的不同作用,分别阐述它们在实验结果中的表达特征和可能意义。我们随后利用GO和KEGG数据库将上述差异基因富集到相应通路中,结合既往相关研究结果,分析这些通路在临床工作或后续研究中可能起的作用。
1 材料和方法
1.1 样本收集
椎间盘髓核样本收集自临床进行手术的椎间盘突出/脱出病人(n=4,年龄53~68岁,2男1女,其中两份样本来自于同一病人的不同节段)和临床因为治疗需要而切除正常间盘的患者,如需要行椎间融合手术的胸椎管狭窄症、脊柱侧弯的患者(n=3,年龄20~48 岁,2 男1女)。上述受试者的IDD进展程度均经过MRI检查,并由改良Pfirrmann退变标准评估分级[10],其中IDD组患者的病变节段经评估分别为5、6、7、8级,非IDD组则均不超过2级(图1)。
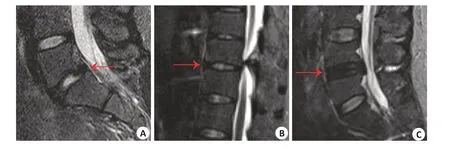
图1 部分入组患者的MRI检查影像Fig.1 MRI images in selected cases.A,B:T2-weighted MR images confirming non-degenerated L5/S1 IVD in a case of spondylolisthesis case and T10/T11 IVD in a case of thoracic spinal stenosis case.C:Obvious degeneration of L4/L5 IVD in a case of intervertebral disc herniation.Arrows indicate the segments for surgery.
IDD组的排除标准包括(1)年龄<40岁或>70岁;(2)合并峡部骨折的腰椎滑脱症;(3)合并其它系统疾病,如严重呼吸系统疾病,心脑血管疾病,消化系统疾病,内分泌系统疾病,泌尿系统疾病,凝血功能障碍等;(4)精神病患者,有吸毒、酗酒不良嗜好者,怀孕妇女;(5)同时参加其他研究者或刚结束其他临床试验研究者。非IDD组的排除标准则为前者的(3)、(4)、(5)两点。本课题研究通过中国重庆市陆军军医大学西南医院伦理委员会审批,受试者均经过知情同意(批准编码:KY2019141)。
1.2 RNA提取及评估
所有样本的RNA均按照制造商指南使用Trizol试剂(Invitrogen)提取,并立即隔离储存于-80 ℃环境。随后RNA样本按说明书指导经Direct-zol RNA miniPrep Kit(ZYMO research)试剂盒分离,RNA 浓度及260/280、260/230吸光度比值则使用NanodropOne分光光度计(Thermo)进行测量[11]。
1.3 高通量转录组测序
本检测项目采用EASY RNA-seq方法完成7个样本的建库,经质控检测后,采用Illumina平台测序[11]。样本测序数据质量的评估标准包括:样本原始测序数据的Q20百分比需超过90%,样本原始测序数据过滤比例大于80%。接下来使用HISAT2比对样本过滤后的测序读长和参考基因组[12],再使用Bowtie2比对样本过滤后的测序读长和参考基因集[13],人类基因的参考序列文件和基因组注释文件均来源GENCODE数据库[14]。通过上述质量检测的样本测序数据则进行接下来的生物信息学分析流程。
1.4 生物信息学分析
首先使用FeatureCounts进行基因原始表达量的统计[15],为了消除样本测序数据量、基因序列长度等差异引起的基因表达量统计偏差,我们采用了FPKM(Fragments Per Kilobase of exon model per Million mapped fragments)方法对样本基因表达量进行标准化[16],并采用小提琴图(Violin Plot)绘制样本基因表达从而标准化数据的分布情况。接着使用R 工具包DESeq2分析组间差异表达基因,在本研究中,显著表达差异筛选标准设定为:P值经过多重校验校正后的值(padj)≤0.05[17]。
我们随后绘制热图对上述实验流程所获得的差异基因进行可视化并利用K-MEANS聚类方法进行分类,从而更好地研究差异基因的不同表达模式[18]。最后我们分别利用GO和KEGG数据库对上述结果进行富集注释,即通过与整个基因组背景相比,分析差异表达基因列表显著富集的相应数据库条目。
1.5 qRT-PCR验证
我们使用GoTaq®qPCR Master Mix 和ABI ViiATM7 Real-Time PCR System验证了组件差异基因的表达。qRT-PCR引物由Primer 5.0软件设计,引物详情见表1。管家基因3-磷酸甘油醛脱氢酶(GAPDH)用作内参对照。完整的热循环体系包括95 ℃3 min、95 ℃15 s以及60 ℃和72 ℃各30 s,共计40循环,熔解阶段由95 ℃30 s和60 ℃1 min组成,运用2-ΔΔCt法计算不同组别目标mRNA间的倍数变化[19],数据以差异倍数的log2值(log2FoldChange)表示,使用R-4.0.4进行分析,采用Wilcoxon符号秩检验进行统计学处理。设定P<0.05为差异有计学意义。

表1 引物信息Tab.1 The primer information
2 结果
2.1 转录组测序及建库
本次研究分别从IDD及非IDD组的RNA文库测得总计84456816和44816454条原始读长,经过筛选,分别剩下78333214和42085446条可用读长,这些可用读长则进一步与人类基因组数据进行对比从而进行下一步的数据筛选(表2)。

表2 测序数据总结Tab.2 Summary of sequencing data
2.2 组间相关性研究及差异基因的获取
差异基因的显著性指标设置为padj≤0.05,将符合此标准的差异基因标注在组间散点图中从而更好地观察它们的散布及表达特征。MA图则用来展示显著差异基因表达水平和倍数变化的散布模式。火山图展示了两组间最具显著性的差异基因(|log2FoldChange|≥1且padj≤0.05)。按上述标准,相对于非IDD样本,共得到了210个上调表达、302个下调表达的显著差异基因(图2)。

图2 组间差异基因的筛选Fig.2 Identification of intergroup DEGs.A:Every point in the figure stands for a gene,X and Y axes separately represents the gene expression level of different groups.Gray dots represent genes with insignificant expression difference,orange dots represent genes which are significantly up-regulated in IDD group compared with non-IDD group,and green dots represent genes that are significantly down-regulated.B:MAdiagram.Each point in the figure represents a single gene,and the X axis represents the average expression level.The Y-axis represents log2FoldChange (IDD/non-IDD expression).C:Volcano plots.Each point in the figure represents a gene,X axis:log2FoldChange,Y axis:-log10(padj).The red dots:DEGs with whose|log2FoldChange|≥1 and padj≤0.05;the green dots:DEGs with whose padj>0.05 and|log2FoldChange|≥1;the grey dots are the non-DEGs.The red-dot-genes were taken into the further analysis.
2.3 差异基因的富集分析
根据分组信息计算不同基因的组内平均表达量,然后用Z评分对其进行标准化。随后对上述标准化平均表达量进行层次聚类,结果以热图的形式展示并基于上述结果进行K-MEANS聚类分析。正常组样本和异常组样本的基因表达趋势不同,而组内的基因表达趋势较一致。另外,表达上调的差异基因虽然数量少于下调基因,但其各基因间的欧氏距离相对较大,上调基因具有较高的离散程度,相对难以富集到一致的通路上(图3)。
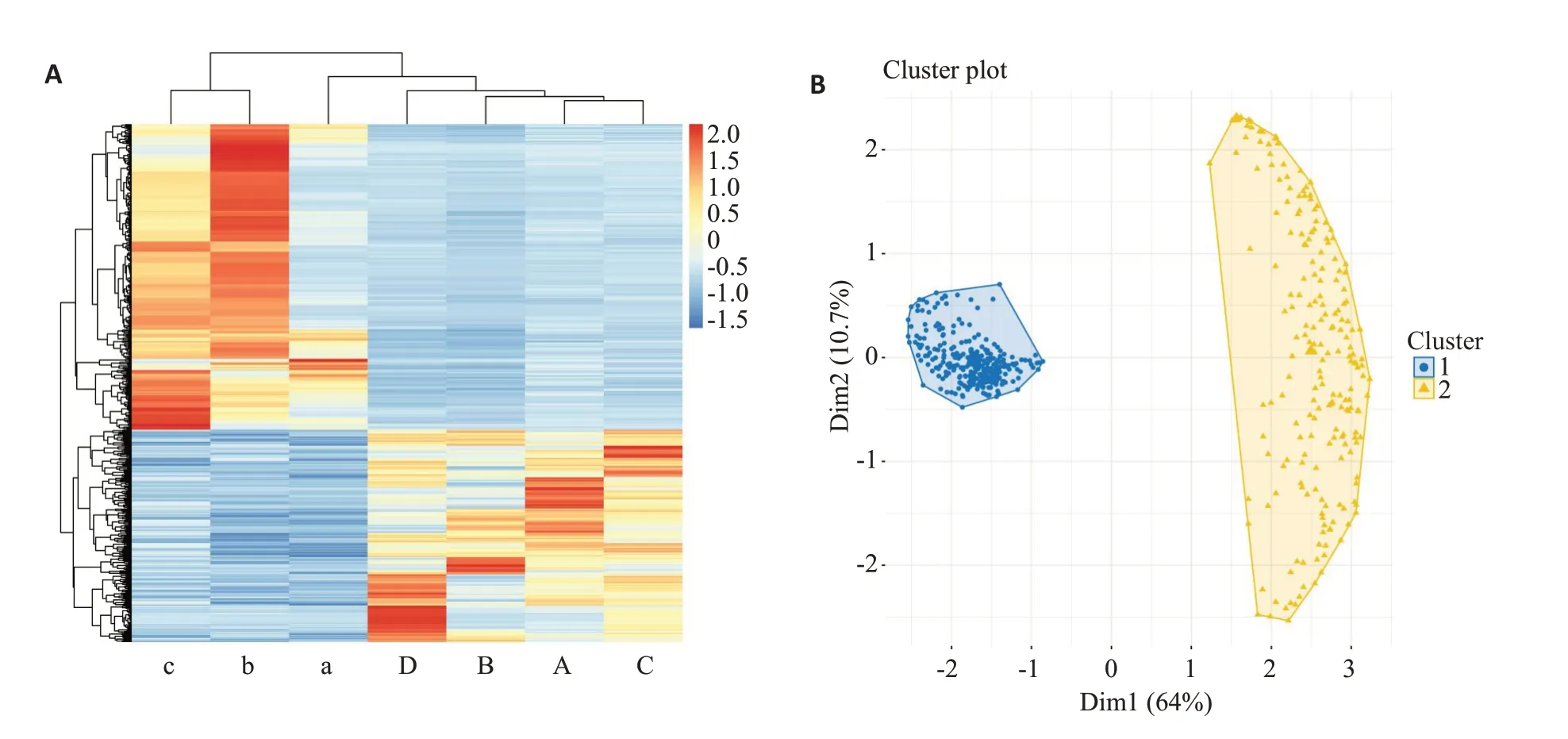
图3 差异基因的富集分析Fig.3 Enrichment analysis of the DEGs in IDD.A:Differential gene cluster analysis diagram.The column represents the expression profile of DEGs,and the row represents the genetic expression quantity among different samples,the left 3 are the samples from non-IDD group,and the right 4 are IDD samples.B:K-means cluster diagram of intergroup DEG set,which also shows the Euclidean distance between genes.
2.4 差异基因的功能富集
分别展示了GO数据库生物学过程、细胞组分、分子功能3个模块以及KEGG数据库的差异基因功能富集结果(图4)。
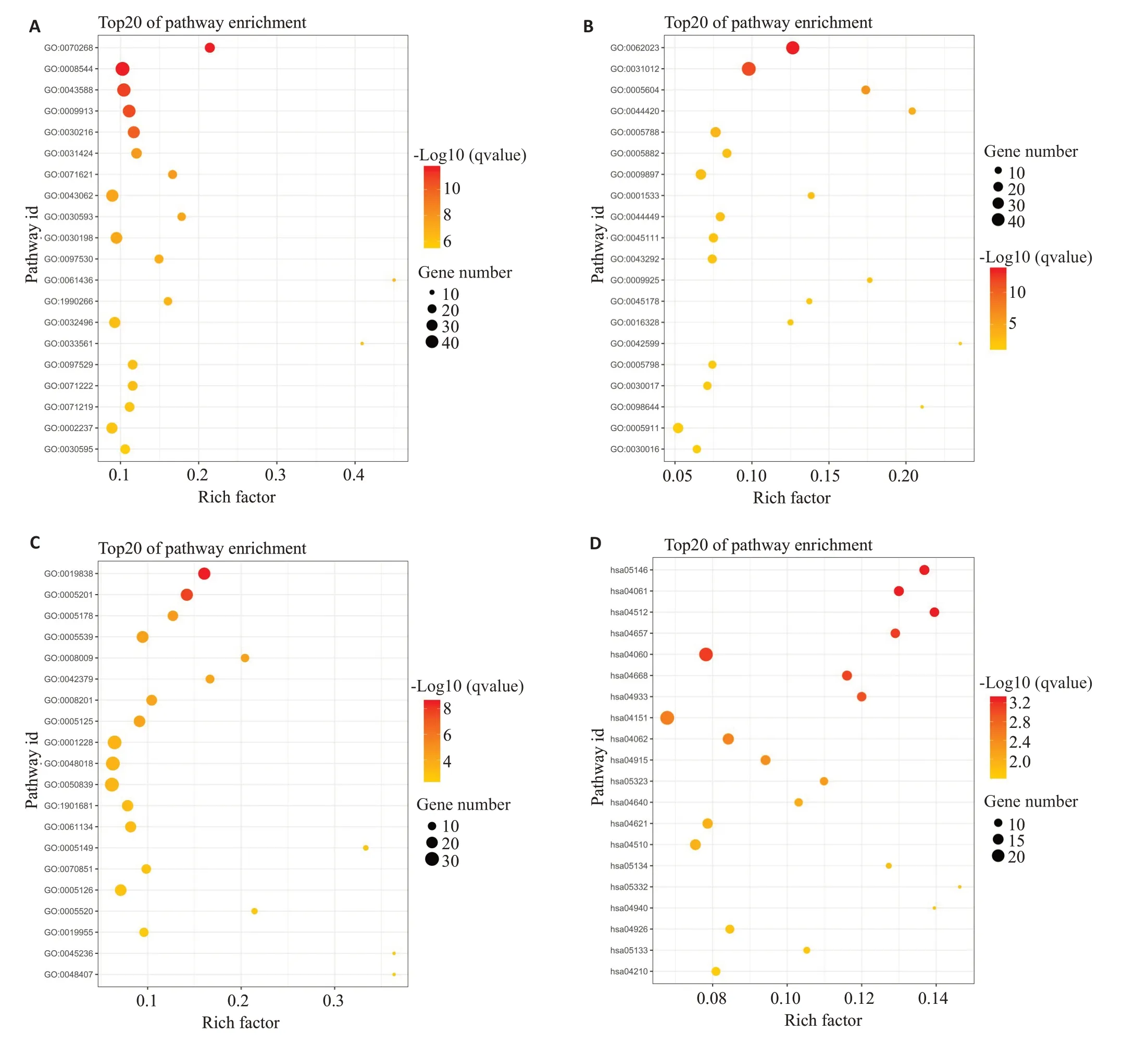
图4 差异基因的GO和KEGG聚类分析Fig.4 GO and KEGG enrichment analysis of the DEGs.A-C:The top 20 terms summarized from the GO biological process,cellular components and molecular function databases.D:Result of KEGG enrichment.
2.5 使用qRT-PCR验证转录组测序结果
根据log2FoldChange绝对值的大小、预期研究价值和既往文献支持选取了13个差异基因作为qRT-PCR的验证目标,分别是FN1、CXCL8、CXCL1、IGFBP6、TNFAIP6、TIMP1、TIMP2、PF4、COL1A1、COL3A1、POSTN、CHI3L2和MMP3,它们在两组间表达具有差异显著性(P<0.001),其中TIMP1、TIMP2和CXCL8的qRT-PCR验证结果经2-ΔΔCt法计算样本组间差异无显著性(Log2FoldChange≤1),其余10个基因具有差异显著性(Log2FoldChange>1)且和RNA-seq相似的表达趋势(图5)。
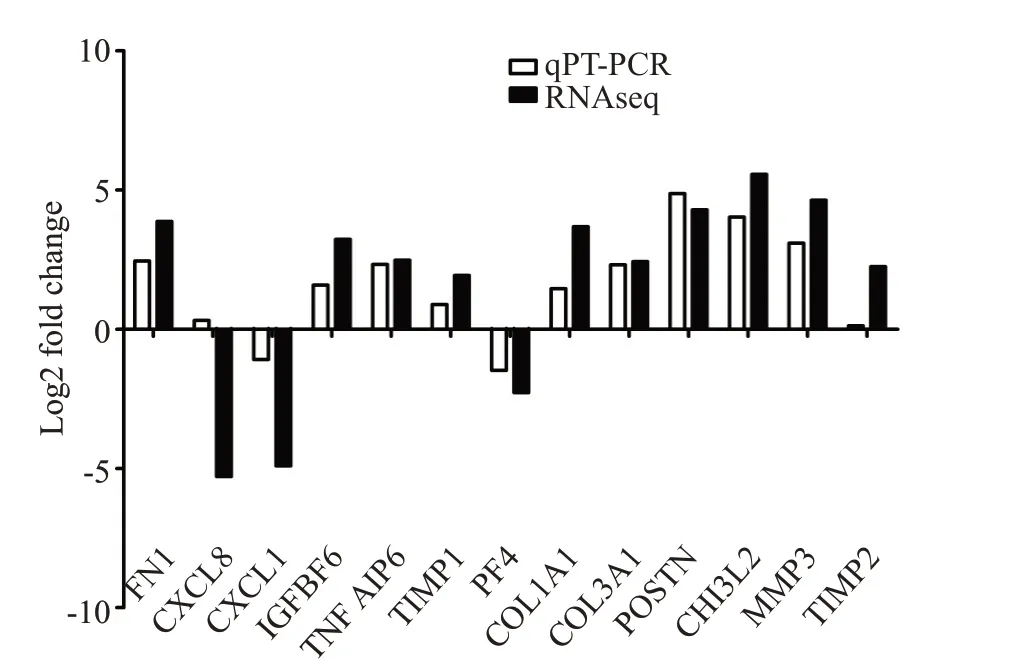
图5 转录组测序结果的qRT-PCR验证Fig.5 Verification of RNA-Seq data by qRT-PCR.Ten of the 13 DEGs show significant intergroup fold change(Log2FoldChange>1),whose expression trends are consistent with the results of RNA-seq.
3 讨论
炎症反应一直是影响包括IDD在内的多种退行性疾病发生发展的重要因素,在本次研究中多种炎症因子也在组间具有显著性差异表达。白介素在机体炎症介导和调控方面具有重要地位,在本次的转录组差异基因中,注意到包括IL32、IL1B、IL2RA、IL7R、IL1RN、IL36RN、IL22RA2、IL1A、IL36G在内的多个白介素相关编码基因对比正常椎间盘组织表达均呈下调趋势,其中既包括具有促进炎症反应的因子,也有IL36RN等炎症抑制剂。这样的结果似乎与既往类似的研究结果不符,一般认为IDD 会伴有大部分炎症介质的表达上调[20]。然而IDD是一个长期的过程,在病程较长的椎间盘突出病例中(距首次症状出现超过半年),炎症反应反而会因退变组织的部分萎缩和降解逐渐减弱。与之类似,C-X-C模体趋化因子配体(CXCL)与C-C模体趋化因子配体(CCL)家族基因,包括CXCL10/9/2/3/1/8 和CCL5/19/22作为人体重要的趋化因子成分之一在IDD组椎间盘样本内表达相对正常组也均呈下调趋势。椎间盘是人体最大的无血管组织,其与血源性的免疫因子接触十分有限,因此,在生理状态下正常的免疫炎症反应很难作用于椎间盘组织。然而在椎间盘突出发生时,髓核通过破裂的纤维环与椎管接触,并且局部血运在生血管相关基因的作用下增加,增大了炎症趋化因子与退变的椎间盘组织作用的可能性。通过炎症因子的作用,突出的椎间盘组织会导致疼痛反应,在少数情况下,其也可能被机体吸收[21]。在本次建立的RNA-seq文库中,多种炎症因子的下调表达可能反映了退变晚期,突出椎间盘无法被机体自我吸收时的基因表达情况。
细胞外基质组分是维持椎间盘髓核细胞正常生态的重要成分,其对维持微环境的含水量及正常代谢环境具有重要作用。胶原蛋白是椎间盘细胞外基质的主要成分之一,已有多个研究提示其不同类型胶原蛋白的含量变化与IDD进展情况具有明显相关性,尤其是Ⅱ型胶原蛋白含量的减少和Ⅰ型胶原蛋白含量的升高被认为是髓核细胞退变的重要标志之一[22]。在本次建库数据中,胶原蛋白编码基因COL1A1、COL4A1、COL3A1、COL4A2、COL18A1和COL12A1均是显著差异基因且对比正常组呈表达升高趋势,而COL2A1虽然未达显著性差异标准,但对比表达下调,与预期结果基本一致,其中部分胶原蛋白成分的可能角色在过去并未被报导。其他细胞外基质组分,包括细胞角蛋白(KRT)等成分,作为正常髓核细胞的表型标志,也呈下调表达[23]。基质金属蛋白酶(MMP)具有强烈的基质降解作用,一直被认为是IDD的重要引导因素,其存在已被证实可能加重包括骨关节炎在内的多种退行性疾病[24]。本次测序证实了MMP3、MMP2在退变椎间盘组织中的上调表达,以及其抑制因子TIMP1/2/3则反射性地表达增强。除上述外,还在差异基因文库中找到了其他可能重要的细胞外基质成分编码基因。几丁质酶3样蛋白2(CHI3L2)缺乏几丁质酶活性,并已被证实在机体对组织重组、损伤和炎症反应方面具有关键地位,其在骨关节炎发生进展中调控软骨再生和Ⅱ型胶原纤维蛋白稳态的作用尤其值得重视[25]。近期有学者指出,它的同源基因CHI3L1可能通过AKT3信号通路在IDD进展过程中起保护性作用,但其中的具体调控机制尚不明确[26]。骨膜蛋白(POSTN)是一类广泛存在于骨骼、皮肤、韧带等人类结缔组织中的细胞外基质分泌蛋白[27],体外实验已证实其可与MMP13基质金属蛋白酶协同作用从而参与骨关节炎进程中的软骨基质降解过程[28]。另有研究者通过免疫学和蛋白组学实验发现POSTN在退变椎间盘细胞中的异常激活,但未进行进一步的探究[29]。以上2种细胞外基质构成组分基因在本次差异对比中上调表达且展现了很高的差异度--其log2FoldChange分别为5.566691773和4.28670097,位于所有差异基因的前10。
众多的生长因子广泛参与人机体的各项生物过程中,椎间盘组织细胞的代谢调控也不例外。既往已有文献指出,包括胰岛素样生长因子(IGF)和转化生长因子(TGF)在内的多个生长因子家族基因变异可能与IDD有关[30]。本次测序发现,退变组织中的胰岛素样生长因子结合蛋白(IGFBP)4/5/6/7对比未退变组织均有不同程度地表达上调,然而与此有所矛盾的是既往研究结果中IGFBP5被认为主要在椎间盘组织中发挥促细胞增殖、抑制细胞凋亡的作用,且往往在IDD进展过程中表达逐渐下调,而全外显子测序结果则猜测IGFBP6的变异与IDD有较高的相关性[31],或许IGF家族及其受体间存在更复杂的信号通路促进或抑制IDD的进展。TGFβ是人体生长发育过程中最活跃、作用最为广泛的信号通路中,各项体内体外实验已证实相当数量的编码基因通过与该通路复杂的交互作用从而影响椎间盘的微环境变化[32]。
随后对上述富集到的所有差异基因进行了GO和KEGG数据库功能富集分析。GO数据库可以从3个方面--生物学过程,细胞组分和分子功能--分析两样本组间差异基因,从而帮助了解IDD在不同生物学层面上的影响因素,从而更加具体地制定下一步研究计划或提出针对性临床诊疗方案。本次主要分析了在各个模块富集基因占比位于前20的通路。在GO数据库生物过程模块的富集结果中,约一半的通路与上皮细胞发育及角化相关,余下的通路则主要包括炎症趋化和细胞外基质合成,综合各通路中富集到的具体差异基因,前者成为差异通路可能与椎间盘中丰富的纤维成分和NP细胞的类上皮细胞表型在IDD中的异常改变有关[33]。细胞组分方面,细胞外基质相关组分无论是在富集度还是基因差异显著度方面都位于前列,而其余通路则包含了各类常见细胞器。综上,这两个模块的富集结果与既往研究结果基本一致,并再次强调了细胞外基质、胞外胶原蛋白成分和炎症因子在IDD发生进展过程中所起的重要作用,后续研究仍可围绕这些成分展开。近来,分子功能越来越成为多种肿瘤、遗传病的研究重点,因此,本研究期望GO数据库的分子功能富集分析模块能带来更细致的答案。在所有富集到的通路中,生长因子结合(GO:0019838)和细胞外基质结构构成(GO:0005201)的显著度远高于其他通路。此前我们已描述了生长因子家族在IDD发生、发展甚至在抑制其发展方面的复杂作用。另外,基于椎间盘细胞质量较少,富含水分和胶原蛋白的环境,细胞外基质对维持NP细胞稳态、负荷外界机械力不可或缺[33]。其余富集到的差异基因通路还包含了多种趋化因子、整合素、肝素与相应受体的结合过程等。KEGG数据库更注重分析基因产物在细胞中的代谢途径以及这些基因产物功能,在本次的分析结果中,不仅IL-17、TNF、PI3K-Akt等对IDD具有调控作用的经典信号通路显示显著性富集作用,而AGE-RAGE和部分糖尿病相关通路也有一定差异性。令人意外的是KEGG富集通路中差异显著性最高的2条分别为阿米巴病(hsa05146,Amoebiasis)和病毒蛋白与细胞因子及其受体相互作用(hsa04061),此外,军团菌(hsa05134,Legionellosis)和百日咳(hsa05133,Pertussis)通路也被差异基因显著性富集,暗示IDD与外界病原体感染有关。然而,IDD作为一种退行性疾病,学界公认其发生发展与年龄增长、机体功能衰退及外界负荷增大有关,一般不会认为病原体对IDD的发生进展会起到首要作用。既往有少数研究指出病原体隐形感染可能会推动IDD进展,但缺乏更多体内实验证实,本次的发现可能为类似观点提供了新论据[34]。
本研究有如下的缺陷:(1)样本量较小,无法很好地更好地设置生物学重复并排除个体间差异。另外,入组患者间,包括年龄、性别、不同的椎间盘节段等也没有很好地统一。造成样本量不足的主要原因是临床上正常椎间盘样本的获取困难--随着诊疗方案和手术技术的进步,非IDD手术患者都越来越少地选择椎间盘摘除术。下一步本课题组可能会使用动物模型来部分弥补该缺陷;(2)RNA-seq技术尽管可以精确快速地获得某一物种特定细胞或组织在某一状态下的所有转录本的集合,从而深入研究mRNA和非编码RNA在IDD进展过程中的变化情况,但其分析仅局限于某一特点时间点,再加上很难确保入组病人处于完全相同的IDD进展阶段,因此可能会带来额外的误差;(3)手术及手术取样时往往是切除整块髓核组织进行RNA提取及后续分析,无法细致地分析髓核内不同细胞群的IDD发生进展因素差异,下一步考虑采用单细胞测序技术,具体区分椎间盘内的各细胞群组,以研究各细胞系对IDD的独特影响;(4)本次研究没有对退变程度不同的样本进行分类差异分析,无法得出IDD进展不同阶段内各自最关键的影响因素,继而寻找IDD进展到不同程度时最佳的治疗方案,这与有限的样本含量和RNA-seq的时限性特点有关。下一步研究本课题组拟加大样本量,并根据改良Pfirrmann分级方法和术中所见病人的突出/脱出程度对入组病人进行分类,以求减少误差,获取更加精确的结论。
猜你喜欢
医学信息(2022年15期)2022-09-09
中国中医药信息杂志(2022年1期)2022-01-13
感染、炎症、修复(2021年1期)2021-07-28
右江民族医学院学报(2021年3期)2021-07-17
中国CT和MRI杂志(2021年5期)2021-04-25
保健与生活(2020年13期)2020-07-24
心电与循环(2020年1期)2020-02-27
当代体育科技(2017年32期)2017-11-27
家庭科学·新健康(2017年10期)2017-10-26
江苏农业科学(2017年5期)2017-04-15
