
牙鲆50K SNP芯片的研制及其在抗病基因组选择中的应用
2021-07-07 07:40周茜陈亚东卢昇刘洋徐文腾李仰真王磊王娜杨英明陈松林
工程 2021年3期
周茜 ,陈亚东 ,卢昇 ,刘洋 ,徐文腾 ,b,李仰真 ,王磊 ,王娜 ,b,杨英明 ,陈松林 ,b,*
a Key Laboratory for Sustainable Development of Marine Fisheries, Ministry of Agriculture, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, China
b Laboratory for Marine Fisheries Science and Food Production Processes, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266373, China
1.引言
单核苷酸多态性(SNP)芯片是一种高质量、便捷的基因分型平台。使用SNP芯片,可同时检测样本中成千上万的SNP,从而实现高通量和高效率的基因组研究和良种选育。SNP芯片已被成功用于多种经济物种的种质鉴定、复杂性状解析、分子标记辅助选育(MAS)和基因组选择(GS)。基因组选择利用全基因组范围内的遗传标记来预测基因组估计育种值(GEBV),并选择具有高GEBV的个体进行育种[1]。基因组选择在育种中的应用非常成功,例如,在许多国家,奶牛育种主要依赖基因组选择和奶牛商业化SNP芯片[2,3]。
在过去几年中,中国已经完成了20多种鱼类的全基因组测序[4]。全基因组序列的获得,促进了养殖鱼类基因组选择技术和SNP芯片的研究和发展。近年来,研究人员研发了多种养殖鱼类的SNP芯片,如大西洋鲑鱼(Salmo salar)[5,6]、鲤鱼(Cyprinus carpio)[7]、虹鳟(Oncorhynchus mykiss)[8]和鲶鱼(Ictaluru spunctatus和Ictalurus furcatus)[9]。然而,目前尚无鲆鲽鱼类SNP芯片及鱼类抗病育种基因芯片的报道。在许多国家,包括中国、韩国和日本,牙鲆是一种重要的水产养殖品种,日本和中国分别于20世纪70年代初和20世纪90年代开始进行牙鲆的良种选育。然而目前,牙鲆养殖的可持续发展面临多种挑战,如种质退化、传染病频发和缺乏优良品种等。因此,迫切需要先进的基因组育种技术培育优良品种,以提高牙鲆养殖的产量和质量。已有一些研究尝试进行牙鲆的良种选育和养殖,例如,鉴定一个抗淋巴囊肿病相关微卫星标记(Poli9-8TUF),并将其应用于MAS[10];通过SNP遗传连锁图谱定位了抗鳗弧菌(Vibrio anguillarum)病相关的数量性状基因座(QTL)[11]等。这些研究有助于我们深化对抗病性状遗传结构的认识。但是,标记数量少,限制了选择育种的效果,而抗病性状是由多个基因控制的复杂性状,因此迫切需要采用基于全基因组范围的SNP的基因组选择进行良种选育[12,13]。
我们使用新一代测序(NGS)技术完成了牙鲆的全基因组测序和组装[14],并基于大规模基因组重测序数据建立了牙鲆抗细菌病基因组选择技术[15]。本研究中,我们设计、研发了一款牙鲆50K SNP芯片“鱼芯1号”,以1099个牙鲆个体的基因组重测序数据为基础,筛选了高质量且信息丰富的SNP研制芯片,并验证了其基因分型效果。当使用“鱼芯1号”芯片作为分型工具开展牙鲆抗细菌病基因组选择时,获得了较高的GEBV估计准确性。因此,“鱼芯1号”芯片在抗病及其他重要经济性状的基因组育种计划中具有应用潜力。任何感兴趣的各方都可以公开获得“鱼芯1号”芯片。
2.材料和方法
2.1.SNP鉴定
“鱼芯1号”芯片的SNP位点来自1099个牙鲆个体的全基因组重测序数据,其中包括Liu等[15]报道的931个个体和本研究测序的168个个体(NCBI SRA登录号SRP253464)。简而言之,从鳍条组织中提取基因组DNA,根据标准方法(Illumina公司,美国)构建双端测序文库。重测序原始短序列在Illumina HiSeq 2000测序平台上产生,然后使用QC-Chain工具进行质量过滤[16]以去除低质量序列、接头序列和不明核苷酸(N)等。使用Burrows-Wheeler aligner工具[17]将质量控制后的序列比对到牙鲆参考基因组(NCBI登录号GCA_001904815.2),然后使用GATK软件(默认参数)[18]预测SNP,以最小比对质量值20、SNP质量得分20和碱基质量得分30等参数进行SNP质量过滤,获得初始SNP集。
2.2.SNP筛选
通过多个步骤及参数对初始SNP集进行筛选。首先,使用PLINK(v1.07)计算最小等位基因频率(MAF)和缺失率[19],去除MAF≤0.05和缺失率≥0.1的SNP;使用VCFtools(v0.1.14)[20]-hwe参数检测哈迪-温伯格平衡(Hardy-Weinberg equilibrium, HWE),并删除严重偏离HWE(p< 0.01)的SNP;将过滤后的SNP及其上下游35 bp侧翼序列提交至Affymetrix Axiom®myDesign GW生物信息分析流程(Thermo Fisher Scientific Inc.,美国)进行探针设计。在该流程中,每个SNP被分配一个p-convert值(介于0和1之间),表示给定的SNP在Affymetrix Axiom芯片系统上转换为可靠SNP位点的概率,该值综合考虑了SNP序列、结合能、预期的非特异性结合程度以及与多个基因组区域的杂交情况等。根据流程中的p-convert值和其他一些质量控制指标,SNP被分类为“recommended”[p-convert值大于0.6,无干扰多态性(wobble)和polycount = 0]、“not recommended”(p-convert值小于0.4,或者无wobble大于等于3,或者polycount > 0,或者重复计数大于0)、“not possible”(在给定的链上不能构建探针来分型该方向上的SNP)和“neutral”(其他)。仅保留“recommended”或“neutral”类SNP的探针以供进一步分析。同时,要求候选SNP的侧翼序列没有其他变异或重复元件,侧翼序列的GC含量为30%~70%。
在此基础上进行进一步过滤,以确保SNP在整个基因组中分布均匀。我们排除了大多数A/T和C/G类型的SNP,因为这些标记在Affymetrix Axiom芯片平台上占据的空间是其他类型标记的两倍。将最终选择的SNP探针与2000个DQC探针(阴性对照)锚定在芯片上。最后,我们使用SNPeff(v4.2)预测芯片上SNP对牙鲆基因功能的潜在影响[21]。
2.3.SNP芯片分型效果评估
为了评估“鱼芯1号”芯片的分型效果,我们对168个牙鲆个体进行了基因分型,包括从基因组重测序样品中随机选择的96个(用于初始SNP集发掘)和从基因组选择参考群体中随机选择的72个[15]。
从每个样品中提取基因组DNA,并根据Affymetrix Axiom®2.0检测方案进行标记,最终DNA浓度为50 ng∙µL−1,体积为10 µL。DNA杂交和芯片扫描在Affymetrix GeneTitan芯片系统(Thermo Fisher公司,美国)上完成,生成原始数据CEL文件。这些文件被导入Axiom Analysis Suite软件中进行质量控制和基因分型。样品质量控制参数为:DQC值≥0.82、检出率≥0.97、合格样品的百分数≥95%、合格样品的平均检出率≥98.5%(遵循“最佳实践工作流程”),并采用默认的SNP质量控制阈值过滤基因分型结果。
通过信号强度和聚类分析评估SNP的探针转化质量,并计算杂合/纯合基因型的数量。根据这些指标,将SNP分为6类:“PolyHighResolution”(SNP具有良好的聚类分辨率,并且至少有两个样本具有最小等位基因)、“MonoHighResolution”(SNP具有良好的聚类分辨率,但是具有最小等位基因的样本不到两个)、“NoMinorHom”(SNP具有良好的聚类分辨率,但没有样本具有最小等位基因)、“OffTargetVariation”(OTV,被称为脱靶变异集群)、“CallRateBelowThreshold”(SNP的检出率低于阈值,但是其他属性高于阈值)和“Other”(一个以上聚类属性低于阈值)[22]。
为了进一步测试SNP芯片的基因分型质量和准确性,我们从基因组重测序样本中随机挑选了96个个体,并比较了由这两种方法获得的基因型的一致性。
葡萄作为一种喜光植物,所以在促成栽培的过程中进行良好的光照调节是非常有必要的。为了更好的保证棚内葡萄可以得到充分的光照,果农每年都应该为大棚更换新的无滴膜,在这个基础上也可以通过在葡萄藤下设置反光膜和膜下滴灌技术来更好的保证葡萄的光合作用。
2.4.群体结构分析
基于芯片分析获得了168个个体的基因分型数据,我们利用GCTA [23]工具进行了主成分分析(PCA),并绘制了第一个和第二个成分的主成分分析图。
2.5.“鱼芯1号”在抗病基因组选择中的应用
我们在前期工作中,利用全基因组重测序数据,研究了基因组选择技术在牙鲆抗迟缓爱德华氏菌(Edwardsiella tarda)选育中的应用前景[15]。在本研究中,我们使用“鱼芯1号”芯片对72个候选个体进行了基因分型,其中27个个体(包括14尾雄鱼和13尾雌鱼)是16个家系的亲本。使用加权基因组最佳线性无偏预测(wGBLUP)估算GEBV,将双亲GEBV均值作为相应家系的GEBV。估计育种值(EBV)利用包含四代系谱数据的最佳线性无偏预测(ABLUP)进行估算,(G)EBV估计的模型为:
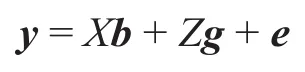
式中,y是表型向量,其中包含个体表型值(0表示在感染实验中死亡,1表示存活);b是固定效应向量(包括均值、不同的感染实验批次以及感染时的年龄);g是随机效应向量;e是随机残差。对于wGBLUP [24],假定随机效应向量服从N(0,G*σg2),其中G*为通过迭代算法得到的加权基因组关系矩阵[25]。对于ABLUP,假设g服从N(0,Aσg2),其中A是具有四代谱系的亲缘关系矩阵;σg是加性遗传方差;X和Z是构造矩阵,分别用于联系表型和固定效应以及表型和个体随机效应。使用R脚本构建加权G矩阵,并在R-ASReml中估算(G)EBV [26]。
由于已经对16个子代牙鲆家系进行了迟缓爱德华氏菌感染实验,wGBLUP和ABLUP的预测准确性可以通过家系GEBV和感染存活率进行评估。将受试者工作特征曲线(AUC)下面积[27]用作衡量wGBLUP和ABLUP预测准确性的指标。为了估计AUC,将16个家系的平均感染存活率(44.33%)作为阈值,高于和低于平均值的家系分别记为1和0。使用R-pROC估算AUC [28]。
3.结果与讨论
本研究旨在研发牙鲆高质量和标准化的SNP芯片,并验证其在基因组选择育种中的应用效果。影响SNP芯片设计和质量的因素很多,如初始SNP集的质量、SNP过滤和筛选参数以及芯片生产技术等。Affymetrix公司和Illumina公司提供了两种最常用的SNP芯片制作平台。两种平台均使用靶标杂交技术检测位点特异性探针,并且探针强度反映了相应等位基因的丰度[29]。在Affymetrix芯片中,指定位置的探针平铺在芯片表面以获得SNP信息,而Illumina芯片则使用微珠固定探针,这些SNP基因分型平台已被广泛应用于遗传学研究。高通量NGS是一种用于鉴定全基因组SNP的有效技术,可用于为SNP芯片筛选SNP。
3.1.测序和SNP预测
我们对168条牙鲆进行全基因组重测序,在质量控制后获得了974.9 Gb的测序数据。将这些数据与来自90 个育种家系的931个个体的测序数据相结合,这931 个个体具有系谱信息并且具有不同的抗病表型[15]。最后,将1099个个体的3.54 Tb测序数据(见附录A中的表S1)与参考基因组进行比对,鉴定了超过4220万个SNP。不同家系个体的大规模基因组重测序使我们能够获得高质量的候选SNP集,这对于芯片的SNP筛选非常有利。
3.2.SNP的筛选和芯片设计
初步鉴定的SNP集使用以下筛选步骤:首先,我们过滤并保留了MAF≥0.05、缺失率小于0.1和未显著偏离HWE(p< 0.01)的SNP。MAF过滤排除了变异频率很低的SNP,缺失率高表明该基因型在群体中的数量有限,HWE过滤排除了由测序错误和自然选择引起的SNP。因此,这些过滤去除了可能影响结果的低质量SNP。过滤后得到一个包含3 410 891个SNP的候选SNP集,这些SNP被提交到Affymetrixin silico探针设计流程,其中959 651个SNP通过了p-convert评估。最后,我们选择了均匀分布于全基因组的48 697个SNP,这些SNP的平均p-convert值为0.684。
3.3.“鱼芯1号”芯片的特征
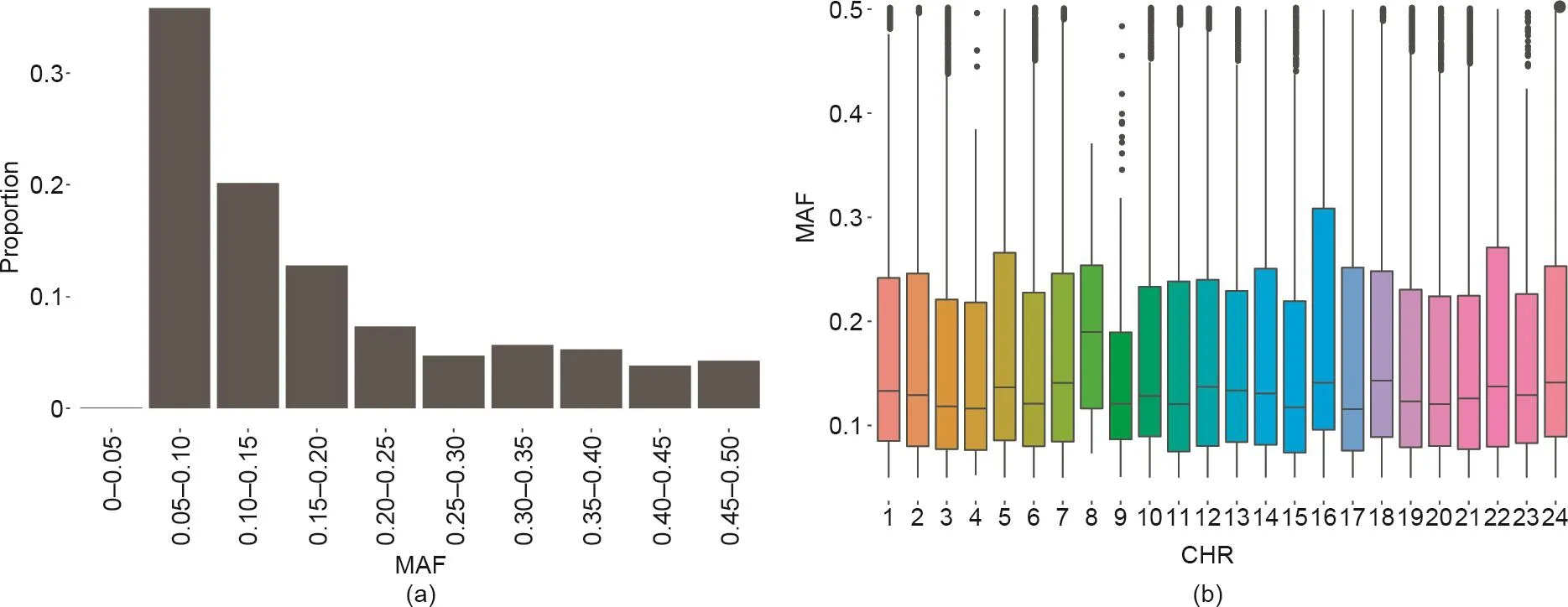
图1.牙鲆“鱼芯1号”SNP芯片中SNP位点的MAF。(a)MAF的比例;(b)MAF在24条染色体上的分布。
为了评估SNP在整个基因组中的分布,我们将芯片上的SNP位点与牙鲆参考基因组进行比对,计算了SNP位点间的距离。我们发现,SNP广泛分布于牙鲆基因组,且相邻SNP位点间具有均匀的间隔,平均间隔距离为9.6 kb [图2(a)]:5125个SNP间距小于6 kb、5175个(10.8%)SNP间距为6~7 kb、6315个(13.1%)SNP间距为7~8 kb、5471个(11.4%)SNP间距为8~9 kb、5546个(11.6%)SNP间距为9~10 kb、6017个(12.5%)SNP间距为10~11 kb、6557个(13.7%)SNP间距为11~12 kb、5964个(12.4%)SNP间距为12~13 kb;累计约96%相邻SNP的间距大于13 kb。这些SNP均匀地分布在整个基因组中,在24条染色体中的平均间距的中位数为9.8 kb[图2(b)]。对于SNP之间距离较大的区域,只有少数SNP符合筛选标准。
对芯片上的所有SNP进行注释,并根据预测效应将其分为不同的类别(表1)。在48 697个SNP中,有26 274个SNP(53.9%)位于基因区,包括外显子、内含子、剪接位点以及基因上游和下游序列的1 kb区域。基因区中两个最丰富的类别是内含子和同义突变,分别包含23 475个和1912个SNP。非基因区SNP包括1684个(3.46%)上游(距离起始密码子1~5 kb)、1754个(3.60%)下游(距离终止密码子1~5 kb)和18 985个(38.99%)基因间SNP。
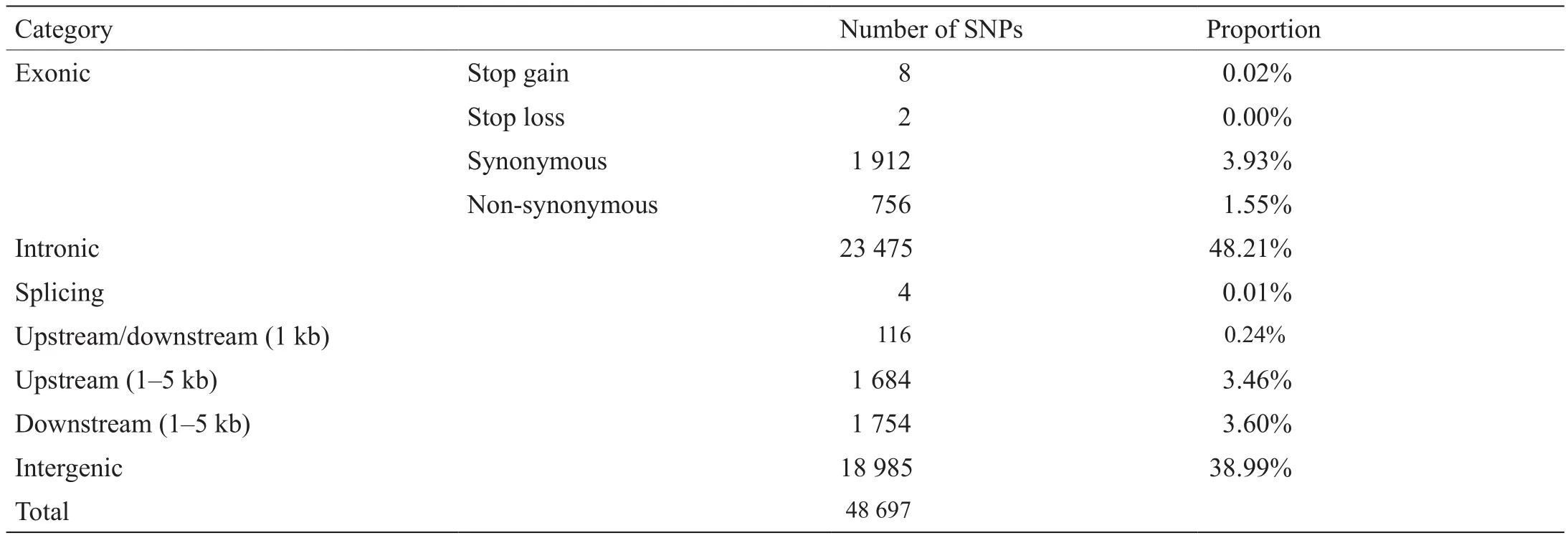
表1 牙鲆“鱼芯1号”SNP芯片上SNP位点的效应类别
3.4.“鱼芯1号”芯片的基因分型效果
采用来自育种家系的168个DNA样本评估了芯片的分型效果,其中166个样本(98.2%)通过了样本质控,检出率阈值为97%。针对基因型检出率、聚类分离、多态性以及芯片与重测序分型的SNP的一致性,评估了“鱼芯1号”芯片的基因分型效果。

图2.牙鲆“鱼芯1号”SNP芯片的位点间距分布。(a)24条染色体上的位点间距;(b)具有不同位点间距的SNP分布。
在“ 鱼 芯1号 ” 芯 片 的48 697个SNP中, 有36 383个SNP(74.71%)通过了所有的质量标准。在这些SNP位点中,有41.07%被归类为多态性的(“PolyHighResolution”和“NoMinorHom”),有33.64%被归类为单态性的(“MonoHighResolution”)。其他位点的基因分型质量较差,聚类效果不好,被分类为“OTV”“CallRateBelowThreshold”或“Others”。检测到较高比例(33.64%)的单态SNP,其中一些可能是SNP发掘过程中的假阳性,或者由于缺乏合适的检测标记而无法对SNP进行有效鉴定。另外,我们使用的168 个个体与重测序进行SNP发掘的群体属于同一群体,因此基因型非常相似;如果对更多的群体进行基因分型,则其中一些SNP可能是多态的。
我们比较了“鱼芯1号”芯片获得的基因型和重测序数据获得的基因型。在包含96个样本的测试中,应用芯片成功对95个样本实现基因分型。在分型成功的SNP位点中,14 899个(41.0%)位点与重测序数据获得的结果一致,4002个(11.0%)、3421个(9.4%)和3162 个(8.7%)SNP的一致率分别为0.95~0.99、0.90~0.95和0.85~0.90。综上所述,70%的SNP的一致率不低于85%,表明“鱼芯1号”芯片和基因组重测序获得的SNP分型结果能够相互验证。
3.5.群体结构的主成分分析
群体结构分析是许多群体遗传学研究的基础。为了评估“鱼芯1号”芯片是否可以检测群体分离状况,我们基于168个个体的SNP进行了主成分分析。根据第一和第二主成分(PC),将所有样本分为两个组(图3),分别对应于我国河北省和山东省的起源/采样地点,证明了“鱼芯1号”具有表征群体结构的能力。
3.6.“鱼芯1号”芯片在基因组选择中的应用
选择育种可以对鱼类的重要经济性状进行遗传改良。我们基于不同的育种家系和迟缓爱德华氏菌人工感染,完成了牙鲆抗病基因组选择技术的研究[15]。为了测试“鱼芯1号”芯片在基因组选择中的应用效果,我们应用芯片对16个随机选择的家系的亲本(共27个个体)进行基因分型,并利用参考群体估算了(G)EBV [15]。其中7个家系的平均存活率是61.13%(命名为抗病家系),其余9个家系的平均存活率为31.27%(命名为易感家系),抗病家系的平均GEBV(2.10)高于易感家系的平均GEBV(1.56)(表2)。由图4可知,wGBLUP的预测准确性高达80%,超过了ABLUP方法(66%),并且与ABLUP方法相比,将SNP芯片和wGBLUP相结合预测育种值的准确性相对提高了21.21%。此外,GEBV与EBV之间中等强度相关(Pearson相关系数为0.70),表明基因组选择方法和ABLUP方法预测育种值的准确性具有差异。我们的结果与已报道的鱼类抗病基因组选择研究一致,即与ABLUP方法相比,基因组选择方法在GEBV估计方面表现得更好,预测准确性提高了13%~52% [30–32]。上述结果表明,“鱼芯1号”芯片可用于牙鲆抗病基因组选择育种。然而,本研究中用于估计GEBV的个体数有限,不能完全模拟牙鲆抗迟缓爱德华氏菌的基因组选择,因此,需要增加个体数目以全面评估“鱼芯1号”芯片用于基因组选择的效果。目前,我们正在努力增加参考群体和候选群体的样本数量,并使用SNP芯片完成基因分型。

表2 16个牙鲆家系感染迟缓爱德华氏菌后的存活率及估计育种值
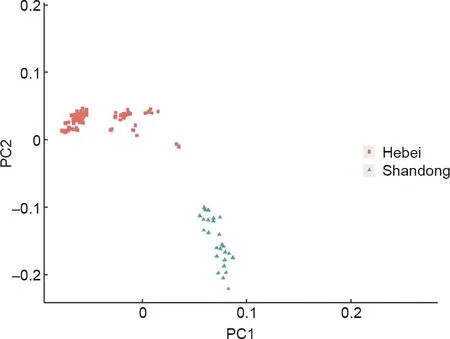
图3.使用牙鲆“鱼芯1号”芯片获得的基因分型结果开展种群结构主成分分析。“Hebei”“Shandong”分别表示在我国河北省和山东省收集的个体。
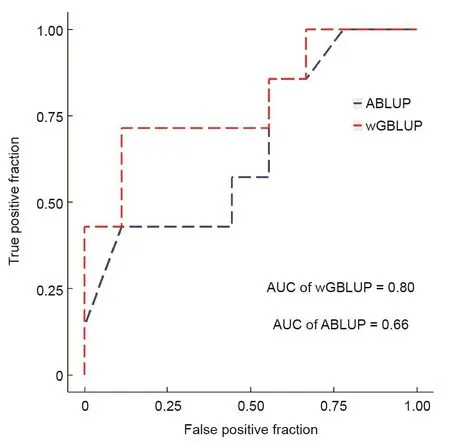
图4.使用受试者工作特征曲线评估wGBLUP和ABLUP对基因组选择的预测准确性。
4.结论
本文报道了牙鲆50K“鱼芯1号”SNP芯片的设计和研发。利用1099个个体的全基因组重测序数据,鉴定了超过4220万个变异位点的起始SNP集;根据MAF、基因组位置和Thermo Fisher Axiom®技术的探针设计建议,选择了48 697个SNP制作芯片。利用“鱼芯1号”芯片,获得了168个样本的高质量的基因分型数据,这些数据用于抗病基因组选择中的效果与已报道的研究一致,预测准确性高于传统基于系谱的BLUP方法。结果表明,“鱼芯1号”芯片适用于重要经济性状的基因组选择,可以为牙鲆基因分型和良种选育提供一个重要的技术平台。
致谢
本研究得到了山东省自然科学基金(ZR2016QZ003)、国家自然科学基金(31461163005)、中央级公益性科研院所基本科研业务费(2020TD20和2016HY-ZD0201)、青岛海洋科学技术国家实验室支持的鳌山科技人才培养计划(2017ASTCP-OS15),以及山东省泰山学者攀登计划项目的支持。
Authors’ contribution
Song-lin Chen obtained the funding, and conceived and instructed the study.Qian Zhou performed the SNP selection and probe design for the SNP array.Ya-dong Chen and Yang Liu prepared the DNA sample.Qian Zhou, Sheng Lu, and Yadong Chen performed the SNP array scanning and analyzed the genotyping data.Sheng Lu performed GEBV calculation.Yang-zhen Li, Lei Wang, and Yingming Yang performed the family construction and bacterial challenging experiment.Wen-teng Xu and Na Wang participated the project managements.Qian Zhou, Sheng Lu, and Song-lin Chen wrote the manuscript.All authors reviewed the manuscript.
Compliance with ethics guidelines
Qian Zhou, Ya-dong Chen, Sheng Lu, Yang Liu, Wenteng Xu, Yang-zhen Li, Lei Wang, Na Wang, Ying-ming Yang, and Song-lin Chen declare that they have no conflict of interest or financial conflicts to disclose.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2020.06.017.
猜你喜欢
北方农业学报(2022年3期)2022-08-08
中国渔业质量与标准(2021年5期)2021-11-20
河北渔业(2019年11期)2019-12-11
现代检验医学杂志(2016年3期)2016-11-15
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
物理实验(2015年9期)2015-02-28
郑州大学学报(医学版)(2015年2期)2015-02-27
中国海洋大学学报(自然科学版)(2014年8期)2014-02-28
