
泪管-耳-齿-指综合征表型与基因特征分析及文献复习
2021-07-06 02:58李霞林垦苏栋高映勤周丽娟刘秀芬马静
中国耳鼻咽喉颅底外科杂志 2021年3期
李霞,林垦,苏栋,高映勤,周丽娟,刘秀芬,马静,2
(1.昆明市儿童医院 耳鼻咽喉头颈外科,云南 昆明 650228; 2.昆明市儿童先天出生缺陷防控研究重点实验室,云南 昆明 650228)
泪管-耳-齿-指综合征(lacrimo-auriculo-dento-digital,LADD),是一种常染色显性遗传疾病,首次于1973年被David W.Hollist等报道并命名,也称为Levy-Hollister综合征。该疾病主要特征为泪道系统发育不全、耳解剖结构和听力异常、唾液系统发育不全、牙齿异常和手指、脚趾发育畸形[1-3]。在后续的相关报道中,又发现了一些可能并存的症状,如面部畸形、肾脏和呼吸系统畸形以及生殖器异常[4]。此病为罕见的遗传性疾病,不同患者之间症状和体征差异较大。该病与其他遗传病综合征也有重叠的症状和体征,为临床诊断该疾病带来困难[5]。
1 临床症状
1.1 泪道系统
表现为泪溢或无泪、泪腺发育不全及结膜炎。由于泪点、泪小管和泪道部分缺失或闭锁,眼泪无法顺利排出,导致泪溢,常引起慢性和复发性眼部感染,甚至发生泪道囊肿。泪腺的发育不全容易造成患者无泪,导致严重的干眼症。这些症状有时单独出现,有时以不同的组合出现。泪腺发育不全和泪道闭锁同时存在,则不会有泪溢的情况[6]。
1.2 耳与听力
表现为听力损失、杯状耳、小耳及其他类型的耳廓畸形。患者听力损失一般为中重度聋,分为传导性或感音神经性聋。这是LADD综合征中对患者影响最大的症状,它的严重程度决定了患者的生存状态。耳廓外形一般为杯状耳,但也存在部分患者耳廓外形为其他的畸形。
1.3 指、趾畸形
不同患者症状不一,文献报道中有拇指的末端指骨重复,三指甲拇指(长手指状的拇指,有3个指骨),前轴多指(多个拇指),以及第3拇指和第5拇指斜指、并指、指缺失等多种症状。
1.4 其他
①唾液腺发育异常:一般表现为唾液分泌不足,常常和泪腺发育异常共同发生;②齿:小牙、钉牙、牙髓病及龋齿。患者的牙齿比较特别,与正常人的牙齿形状不同,呈现出小尖牙的外形。唾液分泌不足易导致口腔干燥,患者会有严重的龋齿,牙齿可能会早期脱落。而其他还包括牙釉质发育不全,牙齿萌发延迟等症状[5];③泌尿系统表现为肾脏异常、生殖不全(胚胎生长过程中器官发育失败)或肾硬化(肾脏硬化)、裂孔疝(胃上部通过胃突出进入胸腔)和冠状尿道下裂(冠状沟内口的腹侧和近端错位)[7-8]。LADD综合征患者也有颅内表现,如基底神经节钙化[9]。
我们通过“LADD”“LADD syndrome”“Lacrimo-auriculo-dento-digital”等关键词在Pubmed数据库进行检索,筛选出26篇文献,共47例患者进行相关症状统计,LADD综合征以耳部症状最为多见,肢端异常为最少见。统计结果见图1。

图1 47例患者LADD临床症状统计
2 基因信息
过去LADD综合征以单纯的临床症状及检查结果异常报道的家系及个案为多,综合征表型差异大,与其他综合征重叠,给临床诊断带来很大的困难。随着基因检测的快速发展及临床应用,LADD患者在基因层面得到了确诊。目前可确定成纤维细胞生长因子受体(fibroblast growth factor receptor,FGFR)2、3及成纤维细胞生长因子10(fibroblast growth factor 10,FGF10)异常表达是该病的致病原因[10]。FGF10和FGFR2、FGFR3属于FGF家族成员。至少有22个成员的FGF家族通过结合和激活4个酪氨酸激酶受体(FGFR 1~4)介导多种生物学功能,例如细胞增殖、分支形态发生和组织分化[11]。FGFR1、FGFR2和FGFR3基因的组织特异性交替剪接产生同工型,它们差异性结合特定的FGF配体[12]。配体-受体结合可能激活多个细胞内级联反应,包括ERK/RAS/MAPK,P13K和PLC-γ/PKC途径[13]。
FGF10位于染色体5p12,包含3个外显子,编码的蛋白质包含208个氨基酸。FGF10是上皮中的间充质信号分子,在上皮间质中转换,对组织损伤的修复和胚胎干细胞分化发挥着重要作用。该基因的异位表达可在体外和体内诱导多种类型的腺体,如泪腺、鼻及眼来源的腺体[14]。
FGFR 1~4表现出不同的时间和空间表达模式。FGFR由胞外配体结合结构域(由3个Ig样结构域组成,称为D1、D2、D3,连接D1和D2接头中的氨基酸段和D2中的保守正电荷区组成作为硫酸肝素或肝素的结合位点),一个单跨膜螺旋片段,一个胞内酪氨酸激酶结构域组成。并且FGFR 1~3在D3的C末端一半处另外进行剪接,以生成“IIIb”或“IIIc”剪接同工型。已经表明IIIb同工型在上皮细胞中表达,而IIIc同工型在间充质细胞中表达(https://www.omim.org)。
FGFR2位于染色体10q26.13,包含24个外显子,编码的蛋白质包含821个氨基酸。FGFR2是FGFR亚家族的一员。FGFR2是软骨内成骨和膜内成骨的重要调和剂。FGFR2突变会导致中耳畸形,从而导致传导性听力损失。在Rigueur等[15]的研究中,敲除FGFR 2的小鼠显示听觉大泡、听骨链、砧骨关节附着的致密结缔组织的多重异常。
FGFR3位于染色体4p16.3,包含19个外显子,编码的蛋白质包含806个氨基酸。FGFR3的突变是骨生长的负性调节因子,它能引起各种短肢骨发育不良和颅结节病综合征。Hayashi等[16]的实验提示FGFR3敲除的小鼠,内、外柱细胞无法完全分化,内柱细胞对FGFR3的变化更为敏感,内、外柱细胞缺损可引起感音神经性耳聋。Mansour等[17]所做的动物实验提示Corti器官的支持细胞可能需要FGFR3来维持和形成。一些突变方式提示FGFR3的突变导致的内耳改变可能具有剂量敏感型。
3 基因的相互作用
FGFs与FGFRs的特异性结合对哺乳动物的发育至关重要,FGFR1~3,D3由不变外显子编码Ⅲa编码,以及外显子选择性剪切的Ⅲb(b)和Ⅲc(c)同工型,包含D2和D3的FGFR片段是与FGF配体结合的最小单位。其中FGFR2Ⅲb(b)型即FGFR2b与FGF10特异性结合[18]。
FGF10对于多种器官的发育是必不可少的,在小鼠模型中,敲除FGF10基因和FGFR2b基因的表型是相似的。这表明FGF10充当了小鼠多器官发育中FGFR2b的特异性配体。FGF10以旁分泌的方式调节细胞内的信号活动。旁分泌FGF的作用是通过细胞外,跨膜和细胞内酪氨酸激酶域激活FGFR(约800个氨基酸)来介导的。
泪腺发育过程复杂,该形态发生始于上皮-间质相互作用启动上皮出芽。FGF10介导的FGFR2b信号传导通过刺激腺上皮细胞的细胞前体中的细胞分裂和基质重塑来启动腺形态发生。多项研究表明,通过对FGFR2b和FGF10突变小鼠和无效小鼠进行研究,FGF10/FGFR2b信号转导途径对于分支器官的发育至关重要,包括肺、乳腺、泪腺、胰腺、甲状腺和唾液腺[19]。
在上述统计结果中,有8例患者进行过基因检测,根据检测结果进行分类统计,结果如表1。
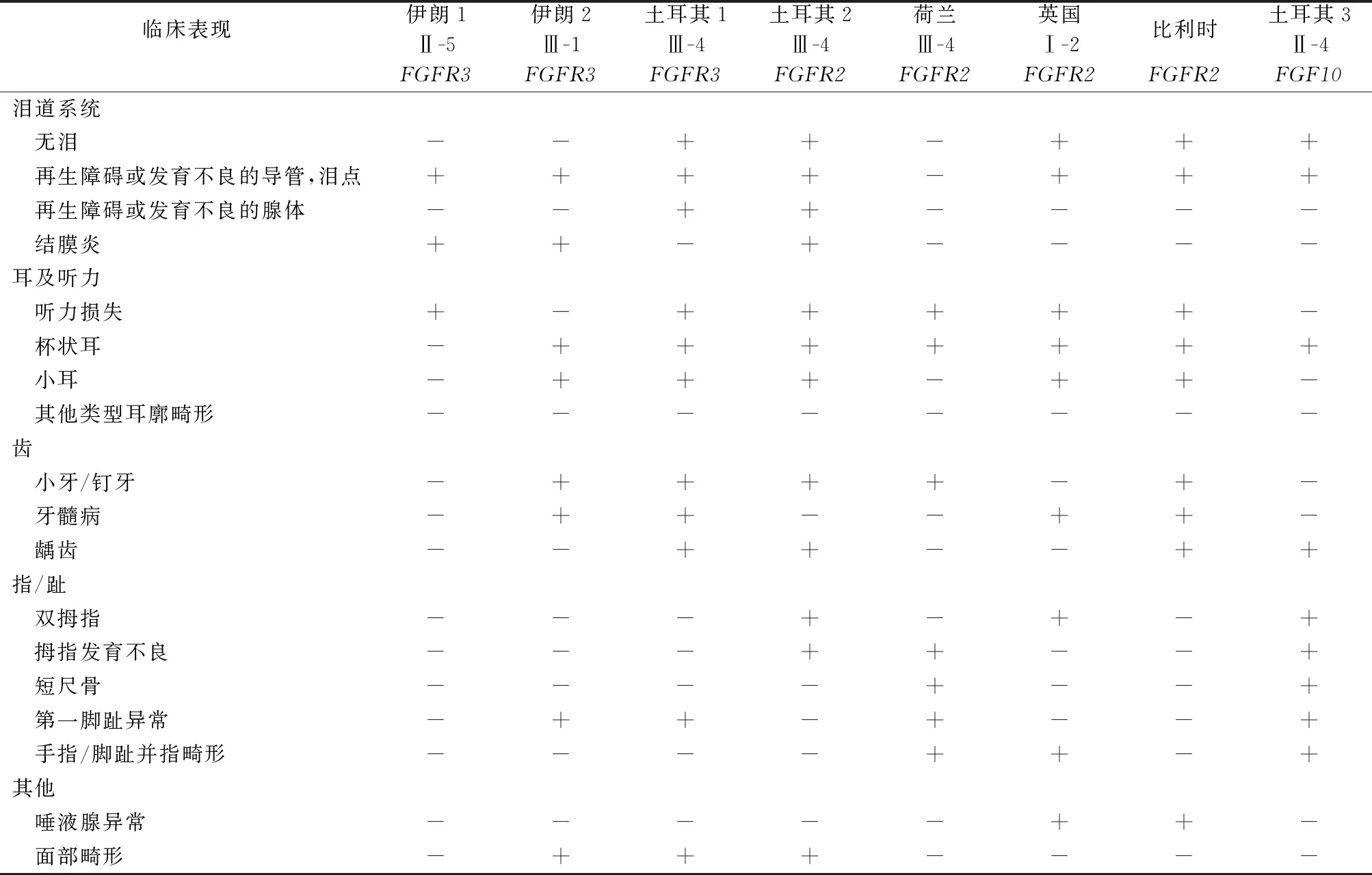
表1 8例患者基因检测与临床表现统计
根据统计结果显示,具有FGFR3、FGFR2、FGF10基因突变的患者,基本都有听力损失,同时存在耳廓畸形的情况,同一家系相同基因位点突变表型有差异,其他症状与各基因间无特殊关联。
4 鉴别诊断
泪腺和唾液腺发育不全综合征(aplasia of lacrimal and salivary glands syndrome,ALSG):其特征为泪腺和唾液系统的发育不全或闭锁。受影响的个体表现出结膜炎和口腔干燥等症状。此疾病为FGF10突变所致,以上特征与LADD综合征表型重叠,相似的症状使得两个疾病难以鉴别。有学者认为ALSG可能与FGF10重要残基上的错义突变有关,而LADD的致病机制更为复杂,临床表型更多[12]。在一些研究中,发现了FGF10核异位有关的分泌缺陷的新机制,在FGF10与FGFR2b结合区域之外的突变会影响核定位及核转运的速度,这可能会影响疾病的表型,但LADD和ALSG疾病的表型异质性说明这不是常见和单一的功能模式[20]。我认为在对两个疾病进行鉴别时,临床表型更多的患者(特别是具有听力损失的患者),诊断为LADD可能性更高,若同时具有FGFR2或FGFR3突变的患者,应诊断为LADD。在将来对具有FGF10突变的患者进行FGF10的鉴定,将ALSG与LADD中的突变进行对比,研究其差异性,将进一步阐明基因型与表型之间的关系,以及他们如何影响FGF10信号的传导,可提高诊断的准确性。
鳃耳肾综合征(branchio-oto-renal syndrome,BORS):主要表现有耳聋、耳前瘘管、鳃裂异常及肾脏畸形,次要表现有外耳形状异常(耳前凸及杯状耳)、中耳畸形、内耳异常、面部不对称及味觉异常等。鳃耳肾的主要致病基因有EYA1、SIX1、SIX5和SAL1、SHARPIN、FGF3、HOX等。BORS因部分临床症状与LADD重叠,在诊断上容易误诊。当患者具有听力损失、耳发育异常及肾脏问题时,根据临床临床表现难以鉴别两个疾病。采用高通量测序,根据检测到的基因突变的结果,若有与上述综合征相关的基因突变,则可进行精准的诊断[5]。
先天性缺指/趾-外胚叶发育不全-唇腭裂综合征(ectrodactyly ectodermal dysplasia and clefting syndrome,EEC):是一类以先天性缺指/趾、并指/趾或手足裂,外胚叶发育不全和伴或不伴腭裂的唇裂三联征为主要临床表现的综合征型唇腭裂,EEC个体可能表现出与LADD表型重叠的特征,包括泪管异常、慢性结膜炎、牙髓病或小牙[20]。在临床中应详细询问病史,仔细进行体格检查,记录每一项异常体征,有条件的情况下,应进行基因测序。ECC为常染色体显性遗传疾病,致病基因为TP63。根据基因检测结果,可对两种疾病精准鉴别。
5 讨论
LADD综合征是罕见遗传病,病症累及多系统,常见泪道闭锁、泪腺、唾液腺等腺体发育不全,牙齿异常,影响外耳、中耳、内耳发育,导致听力损失,以及手指脚趾畸形。另一些少见的症状大多为偶发,无典型特征。目前已知的该疾病致病基因为FGF10、FGFR2、FGFR3。
FGF10主要影响腺体的发育,FGFR2经动物实验证实影响中耳发育,FGFR3敲除在动物实验中表现为影响Corti器的柱细胞发育。FGF10与FGFR2b胞外配体结构域特异性结合,启动腺形态的发生。据Rohmann的研究,在LADD中鉴定出的突变都位于FGFR2与FGFR3的酪氨酸激酶结构域中。在其他疾病中突变则大多位于FGFR的胞外结构中[9]。这提示我们,在FGFR2与FGFR3中鉴定出的突变,应关注其位于编码的蛋白质结构的位置。尽管目前对该疾病的致病机制研究仍不理想,但是基于这些基础,可以进一步揭示致病机制。根据对文献的回顾分析,LADD综合征以耳部情况最为多见,不同基因与症状之间并无特殊关联,但共同的表现仍以耳部情况多见,提示我们在临床中应注意耳部情况,特别是听力的损失,早发现、早干预、早治疗。另外即使同一家系,基因突变位点相同,表型也有差异,提示我们该疾病具有遗传异质性的可能。但因该疾病较为罕见,基因检测的病例数量较少,统计结果可能与实际情况存在一定出入。目前,二代测序技术已成熟,临床应用广泛。在详细分辨记录临床表型的基础上,应使用基因测序,发现致病基因,进一步明确基因与表型之间的关系。另外,通过测序,发现基因不同的突变位点,可丰富遗传信息库。针对这些位点进行致病机制的研究,LADD综合征的致病机制将会更加明确,对患者的治疗将更有针对性,真正造福患者。
猜你喜欢
中国中医眼科杂志(2023年1期)2023-01-16
中国医疗美容(2021年9期)2021-11-02
家庭医药(2021年7期)2021-07-23
时代英语·初中(2018年2期)2018-05-14
时代英语·初中(2018年2期)2018-05-14
时代英语·初中(2018年2期)2018-05-14
时代英语·初中(2018年2期)2018-05-14
现代园艺(2017年21期)2018-01-03
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
