
铁死亡在肿瘤中的研究进展
2021-07-06 01:50孙奋勇
同济大学学报(医学版) 2021年3期
徐 畅,黄 楠,孙奋勇
(同济大学附属第十人民医院检验科,上海 200072)
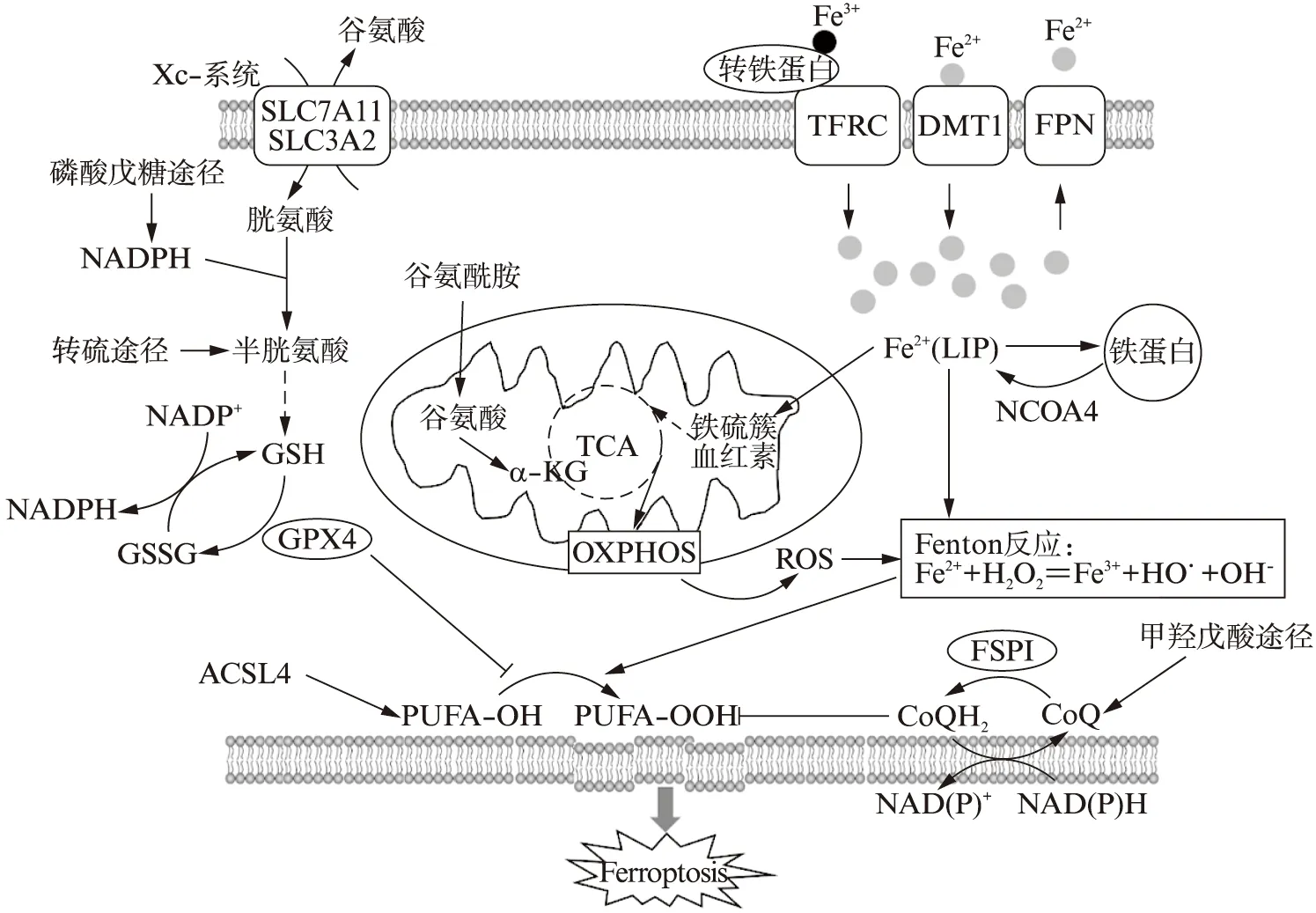
图1 肿瘤铁死亡调控示意图Fig.1 Schematic diagram of tumor ferroptosis regulation
1 肿瘤相关的铁死亡调控
1.1 代谢调控
铁死亡在本质上是铁、活性氧(ROS)、PUFA导致的代谢功能紊乱,肿瘤细胞对铁死亡的敏感性在很大程度上取决于铁代谢、脂代谢、能量代谢等途径的调控。
1.1.1 铁代谢 铁代谢包括循环中的铁稳态和细胞内的铁平衡。摄入的铁经十二指肠上皮细胞吸收,或衰老损伤红细胞中的铁由巨噬细胞吞噬利用,均通过唯一的细胞游离铁出口——膜铁转运蛋白(FPN)输出到外周循环。FPN的表达受肝脏HAMP基因转录产生的铁调素(Hepcidin)抑制[12]。在乳腺癌细胞中,由prominin2-多囊泡体-外泌体介导也可直接将铁蛋白运出细胞[13]。针对循环中铁的不同存在形式,细胞表面含有相应的转运载体,由于细胞内外的pH差异使结合铁以二价形式释放,构成胞质不稳定铁池(LIP)。部分铁以铁蛋白的形式储存,部分经线粒体铁蛋白(MFRN)转运进入线粒体,用于铁硫簇和血红素的生物合成,作为三羧酸循环和线粒体呼吸链的重要辅因子,参与许多关键的生命活动过程[14]。其中,NFS1作为一种半胱氨酸脱硫酶参与线粒体铁硫簇的合成,缺失会引起铁饥饿反应,进而增加肺癌细胞对铁死亡的敏感性[15]。同时,转铁蛋白受体1(TFR1)、二价金属离子转运蛋白1(DMT1)、铁蛋白(ferritin)和膜铁转运蛋白1(FPN1)等主要铁代谢元件,受到铁调节蛋白-铁反应元件(IRP-IRE)和低氧诱导因子-缺氧反应元件(HIF-HRE)两个系统的转录后调控[16]。在三阴性乳腺癌中观察到HIF1α和HIF2α的激活与SLC7A11高表达相关[17]。
由核受体共激活剂4(NCOA4)介导溶酶体对细胞内铁蛋白进行自噬性降解,释放游离铁,引起氧化损伤,这种选择性自噬过程被称为铁自噬(ferritino-phagy)[18]。研究发现,抑制溶酶体功能[19]或沉默NCOA4[20-21]都能抑制肿瘤细胞的铁死亡。BECN1(Beclin1)作为自噬的关键调节剂,在蛋白激酶AMPK激活后触发形成BECN1-SLC7A11复合物,抑制Xc-系统活性,促进铁死亡[22]。
尽管尚不清楚肿瘤发生发展与全身铁水平的关系,但肿瘤微环境或肿瘤细胞中的铁稳态与肿瘤进展有关。肿瘤细胞的无限增殖需要大量铁的支持,在乳腺癌[23]、结直肠癌[24]、前列腺癌[25]、胶质母细胞瘤[26]中,通过调节细胞铁的摄取、存储和输出,表现为铁过载和铁利用率升高,而伴随的LIP增加可能使其对铁死亡更加敏感。
总之,肿瘤细胞通过影响铁代谢元件表达、铁自噬以及线粒体铁利用等过程,改变铁利用率和胞质游离铁水平,从而调节铁死亡的敏感性。
1.1.2 脂代谢 脂代谢相关的肿瘤细胞铁死亡调控主要涉及甲羟戊酸途径和膜磷脂的脂肪酸组成。甲羟戊酸途径是由底物乙酰辅酶A(CoA)依次缩合形成15碳异戊二烯法尼基焦磷酸酯(FPP),除了生成角鲨烯和胆固醇外,还能够进一步反应生成泛醌(CoQ)[27]。其中,角鲨烯[28]和泛醌[29]具有抗氧化功能,而中间产物异戊烯基焦磷酸酯(IPP)则能够有效稳定GPX4翻译所需的硒代半胱氨酸特异性tRNA[10]。值得关注的是,在乳腺癌和骨肉瘤细胞中CoQ的氧化还原酶FPS1是独立于GPX4的重要铁死亡抑制因子[30-31]。同样,硬脂酰CoA去饱和酶1(SCD1)作为催化单不饱和脂肪酸合成的限速酶,能够在卵巢癌细胞中增加内源性抗氧化剂CoQ10来抑制铁死亡[32]。最近的一项研究将细胞色素P450氧化还原酶(POR)鉴定为肿瘤细胞铁死亡磷脂过氧化的正向调控蛋白,促进铁死亡的执行[33]。
目前,该软件的最新版本为2016年2月18日发布的 ERICA Assessment Tool 1.2,可以在网络上免费下载使用。
考虑到PUFA更容易过氧化导致膜不稳定,因此,膜磷脂中饱和脂肪酸与不饱和脂肪酸的相对丰度也会影响铁死亡。脂质重塑过程便是将溶血磷脂酰基转移酶(LPLAT)与磷脂酶A2(PLA2)经过一系列脱酰基和再酰基化,根据不同亚型的LPLAT对脂肪酸底物和磷脂的偏好性,产生多种磷脂[34]。其中,LPLAT3能够增加含PUFA的磷脂比例,进而增加肿瘤细胞的铁死亡敏感性。此外,酰基辅酶A合成酶长链家族成员3和4(ACSL3和ACSL4)也能够调节磷脂的组成。基于不同的底物倾向性,激活不同类型的脂肪酸整合到脂质中[35]。在乳腺癌细胞中,ACSL4介导PUFA掺入磷脂膜来调控铁死亡的敏感性[36],反之,ACSL3利用外源单不饱和脂肪酸置换出膜磷脂中的PUFA,导致铁死亡抵抗。在肾透明细胞癌中,HIF2α也会以ACSL4依赖的方式刺激PUFA特异性富集,促进铁死亡敏感性[37]。
无论是甲羟戊酸途径的抗氧化调节还是磷脂膜脂肪酸的饱和水平,维持一定的抗氧化能力和脂肪酸去饱和状态对于肿瘤细胞的存活至关重要。
1.1.3 能量代谢 能量代谢主要依赖于谷氨酰胺分解、磷酸戊糖途径(PPP)以及三羧酸循环来调节肿瘤细胞铁死亡。一方面,抑制溶质载体家族成员SLC1A5转运体对谷氨酰胺的摄取、阻遏线粒体谷氨酰胺酶GLS2代谢生成谷氨酸,以及阻止谷氨酸通过谷氨酸草酰乙酸转氨酶1(GOT1)生成α-酮戊二酸(αKG),都能抑制铁死亡[38],另一方面,αKG作为三羧酸循环的原料也会影响铁死亡过程[14]。有趣的是,铁死亡的发生似乎依赖于葡萄糖的有效供应,当抑制PPP或是沉默参与PPP的酶——葡萄糖6磷酸脱氢酶(G6PD)和磷酸葡萄糖酸脱氢酶(PGD),能够阻止铁死亡诱导剂Erastin在肺癌细胞中对铁死亡的诱导作用[39]。在肾透明细胞癌中,PPP产生的NAPDH通过还原高表达SLC7A11肿瘤细胞的胱氨酸等二硫化物,维持硫醇氧化还原体系,使其对葡萄糖饥饿敏感并对胱氨酸剥夺诱导的铁死亡抵抗[40]。
1.2 表观调控
表观调控涉及DNA修饰、蛋白修饰、染色质重塑以及非编码RNA等多方面内容,是在不改变遗传信息的基础上,提供表达应用的具体指令。异常的表观遗传调控与肿瘤密切相关,认识铁死亡的表观调控机制有助于开发更加有效的肿瘤治疗手段。
1.2.1 LncRNA长链非编码RNA(lncRNA) 是一类由200多个核苷酸组成的具有低或无蛋白编码能力的非编码RNA,通过直接与DNA、mRNA或蛋白质相互作用,影响细胞增殖分化以及调节性细胞死亡过程。研究表明,染色质调控的lncRNA P53RRA通过与G3BP1相互作用,激活p53进而影响代谢相关基因的转录,促进肿瘤细胞发生铁死亡[41]。非小细胞肺癌(NSCLC)的抑制剂XAV939也是通过lncRNA靶向下调SLC7A11,诱导铁死亡进而抑制肿瘤进展[42]。类似地,肺癌细胞中LINC00336作为microRNA6852的内源分子海绵,促进胱硫醚-β-合酶(CBS)介导的铁死亡抑制作用,降低细胞内铁和脂质ROS的水平[43]。并且lncRNA GABPB1-AS1在erastin诱导的肝癌细胞铁死亡过程中参与调节氧化应激[44]。
关于肿瘤中lncRNA对铁死亡调控的研究仍然十分有限,lncRNA在肿瘤铁死亡中的具体作用机制有待进一步探索。
1.2.2 泛素化调控 BRCA1相关蛋白(BAP1)编码胞核的去泛素化酶(DUB),是葡萄膜黑色素瘤、肾细胞癌、间皮细胞瘤和胆管癌等不同肿瘤细胞系中常见的抑制因子[45]。BAP1能够促进去泛素化酶PR-DUB复合物的形成,去除SLC7A11启动子上泛素化的组蛋白2A(H2Aub),降低SLC7A11的表达[8]。上述过程并不依赖于转录因子NRF2和ATF4的调节,说明H2Aub的动态调控对SLC7A11的表达十分重要[46]。总之,BAP1通过表观调控去泛素化来影响SLC7A11,并由铁死亡介导其部分的肿瘤抑制活性。
另外,赖氨酸120上的组蛋白H2B单泛素化(H2Bub1)是转录激活的表观遗传标记,与糖酵解限速酶丙酮酸激酶M2(PKM2)相互作用调节细胞氧化磷酸化[47]。泛素特异性蛋白酶7(USP7)作为H2Bub1的去泛素化酶,由p53介导促进USP7的核易位并调控H2Bub1,而p53-USP7-H2Bub1能够在erastin诱导铁死亡过程中调控SLC7A11的表达和活性[48]。
关于非组蛋白的泛素化调控研究发现,卵巢肿瘤家族的去泛素化酶OTUB1能够直接与SLC7A11相互作用,调节其稳定性[49]。此外,神经元前体细胞表达的发育下调蛋白4(Nedd4)在erastin诱导后,通过转录因子FOXM1被激活,作为E3泛素链接酶降解线粒体电压依赖性阴离子通道VDAC2/3,调节黑色素瘤细胞对erastin诱导铁死亡的敏感性[50]。
1.2.3 其他表观调控因子 淋巴特异性解旋酶(LSH)在正常细胞发育、代谢和肿瘤进展中具有重要作用[41,43,51-52]。作为一种DNA甲基化修饰因子,在肺癌中由HIF1α介导被c-Myc和Egl9同源物(EGLNs)直接激活,有助于WD重复结构域76(WDR76)募集到SCD1和脂肪酸去饱和酶2(FADS2)基因的启动子,上调其表达,影响脂质ROS和铁的积累[51]。此外,硒(Se)介导的转录适应性调控能够将转录因子激活增强子结合蛋白2C(TFAP2c)和转录因子Sp1募集到GPX4的启动子区域,通过上调GPX4有效阻止铁死亡的脂质过氧化[5]。
1.3 基因调控
肿瘤本质上是各种致癌突变导致的无限增殖和恶性转化,部分癌基因或抑癌基因通过铁死亡发挥作用,这里以经典的抑癌基因p53和Hippo-YAP信号通路为例,介绍基因水平上肿瘤中的铁死亡调控。
1.3.1 p53 p53是重要的抑癌基因,在一半以上的癌症中被突变或失活。其肿瘤抑制活性主要依赖于细胞周期停滞、衰老或凋亡的诱导,通过调控新陈代谢和氧化还原状态也能调节肿瘤的铁死亡[53]。根据p53的突变状态和细胞环境,具有促进或抑制铁死亡的双重作用。
细胞应激时,p53通过抑制SLC7A11转录,减少胱氨酸摄取,促进肿瘤细胞铁死亡。例如,nutlin-3激活p53会触发ROS应激,从而引起骨肉瘤细胞发生铁死亡[7]。对SLC7A11表达的调节依赖于乙酰化p53的DNA结合结构域,带有三个突变赖氨酸的乙酰化缺陷型p53突变体(K117/161/162R,称为p533KR)对铁死亡敏感,限制体内肿瘤生长[54]。p53的错义突变形式,如p53R273H和p53R175H,能阻止NRF2介导的SLC7A11上调,抑制SLC7A11表达[55-56]。除了调节SLC7A11,p53还可以靶向多胺代谢途径有关基因亚精胺/亚精胺N1-乙酰基转移酶(SAT1)来调节肿瘤的铁死亡敏感性[57]。在淋巴瘤中,p53介导的铁死亡依赖于花生四烯酸12脂氧合酶(ALOX12)而非ACSL4,ALOX12的错义突变能够防止PUFA氧化,对erastin或GPX4抑制剂的铁死亡诱导作用是必要的[58]。
在某些条件下,p53还可以负调控铁死亡。例如,野生型p53通过维持p21水平,减少还原型谷胱甘肽(GSH)消耗和脂质ROS的积累,使纤维肉瘤、肾癌和骨肉瘤细胞在胱氨酸剥夺等代谢应激条件下具有生存优势[59]。一致地,在结直肠癌中,缺失p53会阻碍二肽基肽酶4(DPP4)在细胞核中积聚,促进DPP4和NOX1(NADPH氧化酶1)复合物形成,增强脂质过氧化作用和铁死亡[60]。
总之,p53对铁死亡的作用依赖于基因突变情况和氧化应激环境,不同肿瘤中p53调控铁死亡的确切机制有待进一步阐明。
1.3.2 Hippo-YAP 信号通路侵袭转移是肿瘤恶性转化的重要特征,也是难治的主要原因之一,由E- 钙粘蛋白(ECAD)介导的肿瘤细胞间黏附性改变在侵袭转移过程中具有关键作用。研究证实,铁死亡在一定程度上依赖于细胞密度,而ECAD表达水平与细胞融合呈正相关,可以通过肿瘤抑制因子NF2来驱动Hippo信号通路[61]。NF2的激活能够下调E3泛素连接酶CRL4-DCAF1,抑制Hippo通路中激酶LATS1/2的降解[62],进而介导促癌的转录共激活因子YAP磷酸化,消除其核定位,抑制YAP的底物——转铁蛋白受体1(TFRC)和ACSL4等铁死亡相关调控因子,导致铁死亡抵抗[63]。Hippo-YAP通路的异常与多种肿瘤的发生发展密切相关,该途径对铁死亡的调控也为侵袭转移癌细胞的铁死亡敏感性提供了合理的解释。
2 铁死亡在肿瘤治疗中的应用
随着铁死亡调控研究的不断深入,铁死亡诱导剂(ferroptosis-inducing agents,FINs)对于肿瘤治疗的价值日益突显。诱导肿瘤细胞铁死亡,有望解决传统放化疗耐药的问题,将FINs配合新兴的纳米药物、免疫治疗等策略,也为更加有效的肿瘤治疗方案提供了可能。
2.1 铁死亡诱导剂
根据不同的作用机制,将FINs分为以下四类[64]:Ⅰ类的作用是消耗GSH,Ⅱ类直接靶向GPX4使其失活,Ⅲ类通过甲羟戊酸途径消耗GPX4和CoQ10,Ⅳ类通过增加LIP或氧化铁来诱导脂质过氧化,见表1。
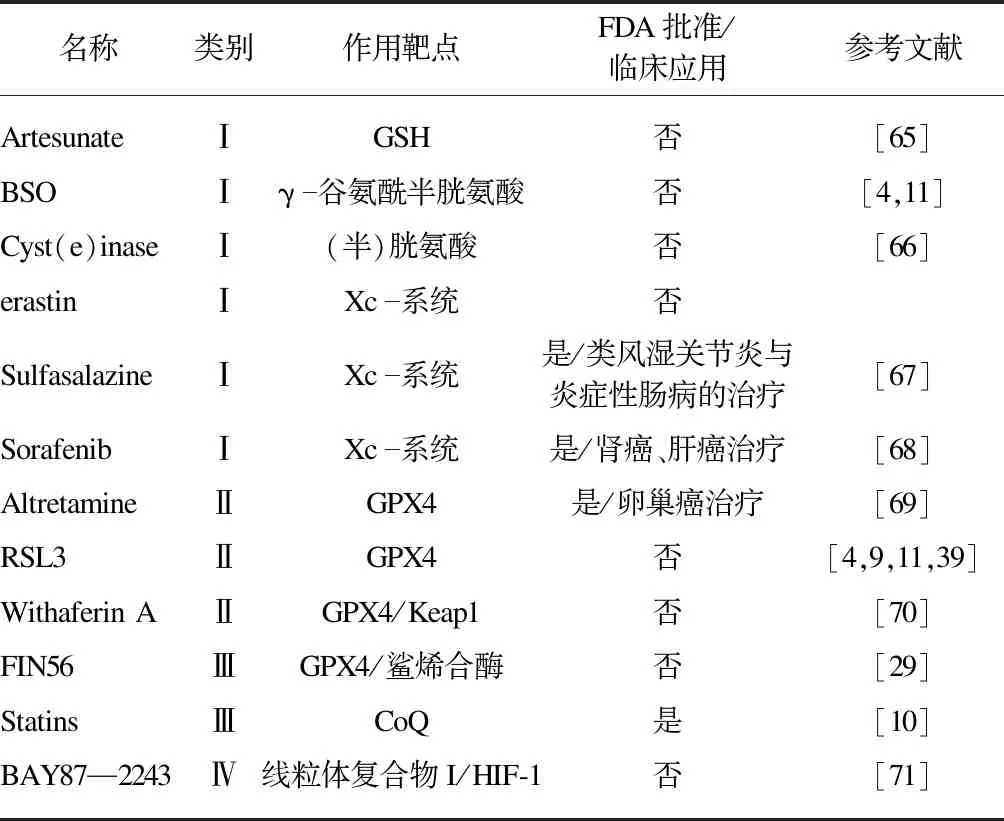
表1 铁死亡诱导剂Tab.1 Ferroptosis-inducingagents
铁死亡诱导是杀伤耐药癌细胞的一种理想方式。研究发现,靶向蛋白激酶和免疫激活治疗抵抗的黑色素瘤细胞表现出铁死亡的敏感性,FINs有助于提高靶向和免疫疗法的功效[72]。同样,锌指E-box结合同源异型盒1(ZEB1)高表达的耐药间叶细胞对GPX4抑制剂或Statins药物诱发的铁死亡高度敏感[10]。并且,癌症治疗中耐药细胞的存活依赖于GPX4[9],使用Ⅱ类FINs withaferin A可以有效杀死抵抗的异质性高风险神经母细胞瘤细胞,在复发率方面较依托泊苷和顺铂更具优势[70]。
铁死亡诱导可以改善常规治疗策略的效果。在高度浆液性卵巢癌(HGSOC)中,高氧化磷酸化亚组由于氧化应激以及潜在的铁死亡作用,化疗敏感性增强[73]。一致地,电离辐射(IR)由ROS和ACSL4介导铁死亡,联合FINs失活SLC7A11或GPX有效增加放疗对抗性癌细胞的杀伤[74]。
2.2 铁死亡与纳米医学
为进一步优化抗癌药物的功效和安全性,纳米医学成为铁死亡应用的新方向[75]。氧化铁纳米颗粒(FePt-NP2)能够提高铁和ROS水平,将顺铂前药装载到FePt-NP2上显著增加了癌细胞对顺铂的敏感性[76]。另外,p53质粒包裹的金属有机网络纳米粒子通过阻碍GSH合成导致铁死亡和肿瘤生长抑制[77]。FDA批准的超小聚乙二醇涂层二氧化硅纳米颗粒在饥饿的癌细胞中介导铁过量摄取,诱导铁死亡并限制肿瘤生长[78]。
利用纳米医学将FINs运至癌细胞,从而避免毒性。例如,withaferin A的纳米颗粒制剂具有增加的通透性和滞留性,在肿瘤处有效积累,抑制神经母细胞瘤的生长[70]。同样,铁死亡诱导剂IKE纳米颗粒也能在弥漫性大B细胞淋巴瘤中蓄积,使毒性降低并促进肿瘤抑制[79]。
尽管铁死亡的纳米疗法改善了肿瘤治疗效果和药物毒理学特性,但由于动物模型和临床实际之间的差异,纳米递药系统的合理设计、规模化制备等仍是目前该领域面临的挑战。
2.3 铁死亡与肿瘤免疫治疗
近年来,铁死亡与肿瘤免疫方面的研究得到快速发展。铁死亡的癌细胞会以自噬依赖性方式释放损伤相关分子模式(DAMP)之一的高迁移率族盒1(HMGB1)[80],使其具有免疫原性,激活固有免疫和适应性免疫。最近的研究表明,巨噬细胞中的铁死亡被一氧化氮通路抑制,为铁死亡调控固有免疫提供依据[81]。胰腺导管腺癌细胞会以自噬依赖性铁死亡的方式释放KARSG12D外泌体,被巨噬细胞吞噬后诱导M2极化过程,抑制其吞噬功能[82]。提示铁死亡在肿瘤免疫逃逸过程中也可能具有关键作用,肿瘤免疫中铁死亡扮演何种角色有待进一步研究。
免疫治疗作为肿瘤领域的研究热点,展现出令人鼓舞的临床疗效。研究表明,免疫治疗激活的CD8+T细胞通过释放干扰素γ(IFNγ)下调SLC3A2和SLC7A11,减少肿瘤细胞摄取胱氨酸,促进其发生铁死亡,而铁死亡又能协同增强T细胞介导的抗肿瘤作用[11]。总之,铁死亡可能是协调肿瘤免疫疗法与常规放化疗的关键枢纽,调控铁死亡对于合理有效的整合免疫疗法与放化疗具有重要意义。
随着铁死亡研究的日益深入,越来越多的证据表明铁死亡在抗肿瘤方面具有积极功效。目前,靶向GPX4的化合物面临着药代动力学和特异性优化等问题,而纳米医学凭借良好的物理特性和肿瘤靶向性,具有低毒性、高安全性的优势,成为铁死亡诱导药物的理想载体之一。由于治疗抵抗的肿瘤细胞普遍存在铁死亡敏感性,现阶段FDA已批准使用Altretamine、Sorafenib等药物作为肿瘤铁死亡诱导剂进行辅助治疗,基于铁死亡分子调控机制的肿瘤治疗应用仍然有着巨大的发展潜力。尽管肿瘤的铁死亡调控研究取得了一定进展,但仍处于起步阶段,迫切需要结合单细胞测序等高通量技术,进一步从代谢途径、表观修饰、基因突变谱等多个角度分析肿瘤中铁死亡的调控机制。同时,相关的基础研究也可着眼于肿瘤中铁死亡的生理功能,比如解析铁死亡在肿瘤免疫中的作用,进而定义对铁死亡治疗敏感的肿瘤特征,积极控制潜在的毒副作用,最终转化推动肿瘤治疗水平的提升。总之,阐明肿瘤铁死亡的分子调控机制有助于推动肿瘤治疗领域的不断突破,为癌症的攻克作出重要贡献。
猜你喜欢
临床外科杂志(2022年8期)2022-11-23
中国药理学与毒理学杂志(2022年7期)2022-10-17
现代仪器与医疗(2022年4期)2022-10-08
生物化学与生物物理进展(2022年8期)2022-08-20
作物学报(2022年10期)2022-07-21
电气技术(2022年6期)2022-06-27
中华骨与关节外科杂志(2021年12期)2021-08-31
现代临床医学(2021年1期)2021-01-26
记者观察·下旬刊(2019年12期)2019-09-10
大陆桥视野·下(2017年8期)2017-09-19
