
锆表面微弧氧化膜1000~1200 ℃高温蒸汽氧化行为研究
2021-07-03 09:25王兴平廖燚钊关浩浩高川力朱明浩金小越徐驰杜建成薛文斌
表面技术 2021年6期
王兴平,廖燚钊,关浩浩,高川力,朱明浩,金小越,徐驰,杜建成,薛文斌
(1.北京师范大学 核科学与技术学院 射线束技术教育部重点试验室,北京100875;2.北京市辐射中心,北京100875;3.国防科技工业核材料技术创新中心,北京102413)
微弧氧化(MAO)是一种在铝、镁、钛、锆等金属表面原位生长陶瓷膜的表面处理技术[1-3]。目前,锆及锆合金在硅酸盐、铝酸盐和磷酸盐等体系电解液中微弧氧化工艺都有报道[3-9]。Zou 等[3]测量出Zr-1Nb合金微弧氧化膜的显微硬度大于 570HV,远高于Zr-1Nb 基体的硬度值。高硬度的ZrO2陶瓷膜有助于提高高温高压水环境中锆包壳管与定位格架间的抗微动磨损性能[10]。Wang 等[11]发现ZrH1.8合金在磷酸盐电解液中制备的微弧氧化膜更致密,阻氢效果优于硅酸盐溶液中制备的ZrO2陶瓷层。研究发现,锆合金在硅酸盐、铝酸盐溶液中制备的陶瓷膜孔洞较多,还夹杂着一些微裂纹[12–14],这不利于提高锆合金在高温高压水环境中的耐腐蚀性能。Sandhyarani 等[15]报道,纯锆在磷酸盐和KOH 的混合电解液中制备的微弧氧化膜比在硅酸盐溶液中制备的膜层具有更好的耐腐蚀性能。此外,磷酸盐溶液中制备的微弧氧化膜降低了纯锆的亲水性,腐蚀电流密度减小1 个数量级[16]。
高温高压水溶液中,锆基材料的腐蚀速率显著增加,因此微弧氧化膜的高温耐蚀性能受到一些学者的关注[17-19]。Yang 等[18]报道,在360 ℃/18.6 MPa 的0.01 mol/L LiOH 溶液中腐蚀94 d 后,微弧氧化膜的腐蚀增重低于Zr-1Nb 合金基体。笔者采用原位高温电化学测试技术,评估了Zirlo 和Zr3Al 锆基材料微弧氧化膜在300 ℃/14 MPa 硼锂缓冲液中的腐蚀行为,发现微弧氧化膜可保护锆基体免受Li+腐蚀,而且均匀致密的内层膜对锆基材料的耐蚀性提高具有关键作用[20-21]。这些表明微弧氧化膜在常温或高温水腐蚀介质中均具有良好的耐蚀性能。
在反应堆失水事故工况(LOCA)下,锆包壳在高温蒸汽环境中剧烈反应,释放大量氢气,引发福岛核事故类似的事件[22-23]。通过调整合金成分和优化加工工艺,开发的M5、Zirlo、E635 和E110 等新型锆合金均未显著提高锆包壳的抗高温蒸汽氧化性[24-26]。因此,作为一种原位生长的锆表面保护涂层,有必要评估锆微弧氧化膜的抗高温蒸汽氧化性能,确定其抗蒸汽氧化的温度范围,探究其在LOCA 工况下的防护作用。
本文利用微弧氧化技术在纯锆表面制备了致密的微弧氧化薄膜。研究微弧氧化表面处理对纯锆在1000~1200 ℃蒸汽环境中氧化行为的影响,分析氧化膜的组织结构、相组成及成分,探讨锆的蒸汽氧化机理和氢扩散机制。
1 试验
试验基材为核级纯锆薄板,用1000#、3000#、5000#水砂纸对规格为10 mm×10 mm×1 mm 的片状试样依次打磨,并冲洗吹干待用。采用双极性脉冲电源(WHYH-100KW)对锆试样微弧氧化处理。加载电压、频率、占空比及处理时间分别为+475 V/–35 V、75 Hz、45%和20 min。磷酸盐电解液组成为6 g/L Na3PO4·12H2O、1 g/L KOH 和20 mL/L 甘油。添加甘油可以抑制破坏性火花的形成,提高微弧氧化膜的致密性。
采用法国塞塔拉姆仪器公司生产的 SETSYS Evolution TGA 热分析仪对锆基体及微弧氧化膜试样进行高温蒸汽氧化测试,测试温度为1000、1100、1200 ℃。试样悬挂在铂挂钩上,将纯度为99.99%氩气以50 mL/min 的速率通入样品室,并以50 ℃/min的升温速率加热至设定温度,然后将相对湿度为50%的蒸汽通入样品室进行高温氧化试验。蒸汽中保温3600 s 后,在氩气环境中自然冷却至室温。每种条件下测试3 个平行样品,保证增重曲线具有较好的重复性。
采用Hitachi S-4800 型扫描电子显微镜(SEM)观察锆基体和微弧氧化试样的表面形貌和截面组织,通过GDA 750HR 型辉光放电谱仪(GDOES)分析微弧氧化膜试样氧化前后的成分深度分布。利用X' Pert Pro MPD 型X 射线衍射仪(XRD)表征氧化前后试样的相组成。通过Carl Zeiss Image A2m 型光学显微镜(OM)观察氧化后的截面金相组织,蚀刻剂选用50%H2O+45%HNO3+5%HF(体积分数)的混合液。另外,用HARKE-SPCAX1 型接触角测试仪分析锆基体及微弧氧化膜的表面润湿性,并采用二液法计算它们的固体表面能。
2 结果及分析
2.1 微弧氧化膜的微观形貌及成分分析
图1 显示了锆表面微弧氧化膜形貌、成分深度分布及XRD 谱图。如图1a 所示,微弧氧化膜呈典型的“饼状”结构,表面残留微小的放电孔洞[27-28]。由于放电区熔体冷淬作用[20],导致表面产生微裂纹(见图1a 插图)。图1b 显示,微弧氧化膜层致密,膜/基界面结合良好。膜厚度约为2.5 μm,内部致密层和外部疏松层分别为2 μm 和0.5 μm。为H 的富集与膜内吸附的水分子有关。微弧氧化过程中,等离子体放电通道内瞬时温度达3000~10 000 K[32–34],因此膜/基界面附近的水分子与锆反应释放出氢气,导致少量H 富集在膜/基界面附近[29]。从图1d可知,微弧氧化膜包含单斜氧化锆(M-ZrO2)和少量的四方氧化锆(T-ZrO2)相,来自锆基体的α-Zr相也被探测到。

图1 锆表面微弧氧化膜的形貌、成分及相组成Fig.1 Morphologies, compositions and phase components of MAO-coated pure Zr: (a) surface morphology, (b) cross-sectional microstructure, (c) GDOES composition depth profiles, (d) XRD pattern.
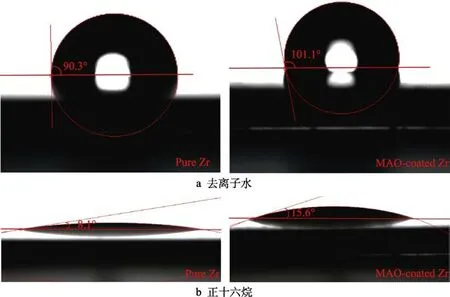
图2 锆基体及微弧氧化膜表面去离子水和正十六烷液滴的接触角Fig.2 Contact angles of bare and MAO-coated pure Zr for deionized water and hexadecane: (a) deionized water, (b) hexadecane
图2 为锆基体及微弧氧化膜表面去离子水和正十六烷液滴形貌。图2a 显示,锆基体表面的平均接触角为90.3°,而微弧氧化膜的接触角为101.1°,表现为疏水性。这表明微弧氧化表面处理提高了锆基体表面润湿角,而且锆基体及微弧氧化膜与正十六烷的接触角分别为8.1°和15.6°(见图2b)。此外,采用Owens-Wendt-Kaelble 方程评估它们的固体表面能[35-36],二液法固体表面自由能计算方程见式(1)和式(2)。
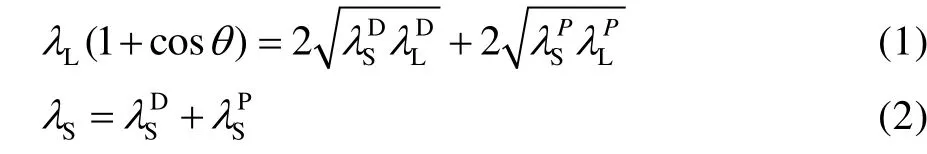
式中:Sλ、和PSλ分别为固体的表面自由能、色散力和极性力;Lλ、和分别为液体的表面自由能、色散力和极性力。研究表明[37-38],水的表面自由能、色散力和极性力分别为72.8、21.8、51 mJ/m2,而正十六烷的表面能、色散力和极性力分别为27.6、27.6、0 mJ/m2。
根据去离子水和正十六烷的接触角数据,计算出纯锆基体及微弧氧化膜的表面自由能分别为30.1 mN/m和28.2 mN/m,锆微弧氧化膜的表面能比锆基体低一些。固体的表面自由能通常与接触角呈负相关关系[39],较大的接触角对应低的表面能。因此,微弧氧化处理后,锆表面自由能降低,其憎水性增强。

图3 锆及微弧氧化膜在25~1250 ℃蒸汽瞬态试验中的氧化动力学曲线(插图为氧化增重速率随时间的变化曲线)Fig.3 Oxidation kinetics curves during the transient test of bare and MAO-coated pure Zr in steam environment at 25~1250 ℃ (The inserted figure is their mass gain rate as a function of oxidation time)
2.2 高温蒸汽氧化特性
图3 为锆及微弧氧化膜在25~1250 ℃蒸汽瞬态试验中的氧化动力学曲线,图中插图为氧化增重速率随升温时间的变化曲线。如插图所示,锆基体及微弧氧化膜的氧化增重速率随蒸汽温度的升高而增加。在25~1250 ℃升温阶段,微弧氧化试样的氧化增重速率略低于锆基体,加速氧化动力学转变温度也未显著提高,约为600 ℃(见插图点A),因此文中的恒温蒸汽氧化试验温度远高于600 ℃,以评估锆试样快速氧化情况。图3 还显示,锆基体和微弧氧化膜的最终单位面积增重分别为4.53、3.73 mg/cm2,表明微弧氧化膜减缓了锆基体的氧化。
图4 为锆及微弧氧化膜在1000~1200 ℃蒸汽中的氧化动力学曲线。通常认为,锆及锆合金高温蒸汽氧化增重符合抛物线规律[40-42]。而图4 显示并非所有温度都遵循抛物线规律,氧化规律同温度和保温时间密切相关。氧化增重-时间关系见方程(3)。

式中:ΔW/A为单位面积氧化增重(mg/cm2);n和Kn分别为氧化速率指数(n=1 或2)和氧化速率常数;t为氧化时间(s)。拟合结果见表1。
图4a 和表1 显示,1000 ℃蒸汽氧化3600 s 后,锆和微弧氧化膜的氧化动力学曲线遵循抛物线规律,微弧氧化膜的Kn值低于锆基体,微弧氧化膜的单位面积增重为7.81 mg/cm2,明显低于锆基体(10.36 mg/cm2)。表明微弧氧化膜在1000 ℃蒸汽中可抑制氧向锆基体内扩散,提高了锆基体的抗蒸汽氧化性能。
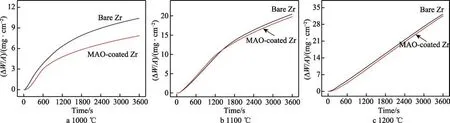
图4 锆及微弧氧化膜不同温度蒸汽中的氧化动力学曲线Fig.4 Oxidation kinetics curves of bare and MAO-coated pure Zr in steam environment at different temperatures
图4b 和表1 显示,1100 ℃的锆及微弧氧化膜的氧化动力学曲线基本遵循抛物线规律。随着氧化时间的延长,增重速率略有下降,这是由于不断增厚的氧化层抑制了氧向锆基体内扩散。1500 s 之前,二者氧化动力学曲线几乎重叠,1500 s 之后,微弧氧化膜试样的氧化增重略低于锆基体。随着蒸汽温度升高至1200 ℃,锆及微弧氧化膜试样的氧化动力学曲线几乎重合,遵循线性规律(见图4c 和表1)。1200 ℃蒸汽氧化3600 s 后,二者的氧化增重达31 mg/cm2。这表明锆表面微弧氧化膜在1100 ℃和1200 ℃蒸汽环境中丧失了保护作用。
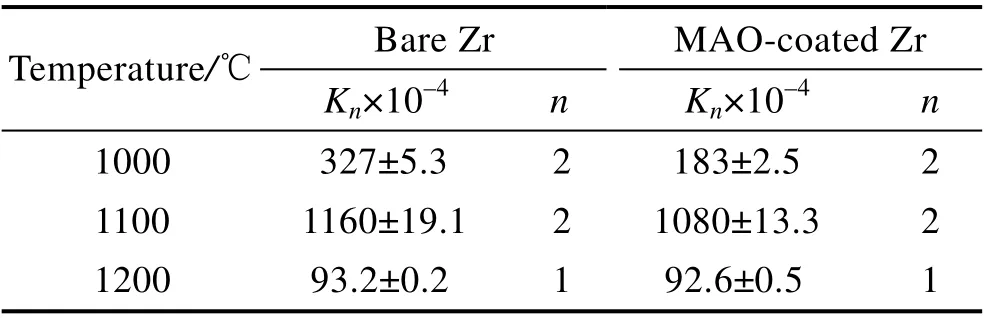
表1 锆及微弧氧化膜在不同温度蒸汽中的氧化动力学参数Tab.1 Oxidation kinetics parameters of bare and MAO-coated pure Zr at different temperatures in steam environment
2.3 蒸汽氧化后氧化层组织和成分分布
图5 为纯锆及微弧氧化膜在1000~1200 ℃蒸汽氧化3600 s 后的截面组织图,不同温度下氧化层和α-Zr(O)层的厚度如图6 所示。图5a 显示,锆基体经1000 ℃蒸汽氧化后,从外到内为ZrO2层、氧稳定的α-Zr(O)层和前β-Zr 相区,ZrO2层和α-Zr(O)层的厚度分别为58.1 μm 和96.6 μm。α-Zr(O)层下方的微观组织呈平行板结构,这一区域被称为前β-Zr 区。图5c和图5e 显示,1100 ℃和1200 ℃蒸汽氧化后,锆基体表面ZrO2和α-Zr(O)层进一步生长,ZrO2层的厚度分别增加至105.1 μm 和156.9 μm,α-Zr(O)层的厚度分别增加至123.3 μm 和251.1 μm。

图5 纯锆及微弧氧化膜在不同温度蒸汽中氧化3600 s 后的截面组织Fig.5 Cross-sectional microstructures of bare and MAO-coated pure Zr after 3600 s steam oxidation at different temperatures: (a,c, e) bare Zr, (b, d, f) MAO-coated Zr

图6 氧化层和α-Zr(O)层厚度随蒸汽温度的变化Fig.6 Dependence of thicknesses of oxide layer and α-Zr(O)layer on the steam temperature
图5b、图5d 和图5f 显示,微弧氧化膜蒸汽氧化后,其截面结构与锆基体氧化层相似。在1000 ℃温度下,微弧氧化膜试样表面ZrO2层和α-Zr(O)层的厚度小于锆基体,表明1000 ℃蒸汽中微弧氧化膜延缓了氧向锆基体内扩散的速度。然而,1100 ℃和1200 ℃蒸汽氧化后,二者表面ZrO2层和α-Zr(O)层的厚度基本相等,表明1100 ℃以上时微弧氧化膜丧失了抗蒸汽氧化能力,这与图4 氧化动力学曲线结果一致。
图5e 和图5f 中还观察到,锆基体及微弧氧化膜经1200 ℃蒸汽氧化后,ZrO2层附近的α-Zr(O)层发生严重局部剥落。高温蒸汽环境中,大量的氧溶解于锆基体,韧性β-Zr 相转变为脆性α-Zr(O)相[43]。在截面样品切割抛光过程中,α-Zr(O)层发生局部剥落,形成剥落区域。
图7 为锆及微弧氧化膜在1000 ℃蒸汽中氧化3600 s 后的成分分布图。图7a 和图7b 显示,大量的O、H 和C 元素富集在0.5 μm 的氧化膜表层,这归因于蒸汽氧化后表面氧化层比原始微弧氧化膜更易吸附大气中的污染物[44],同时与GDOES 成分测量时初期放电不稳定有关。图7b 还显示,H 和C 元素的相对强度从外到里依次递减,来自电解液的P 和Na 元素仍富集在初始微弧氧化膜区域,但相对强度减弱。蒸汽氧化后,原始微弧氧化膜中膜/基界面附近H 峰消失。此外,1000 ℃蒸汽氧化后,锆及微弧氧化膜试样中的氧扩散深度分别达58.1 μm 和31.4 μm,而H 元素仅富集在厚度约3.0 μm 的氧化层中,并未扩散进入锆基体及蒸汽中新生的内层氧化膜。杜培[45]和Cathcart[46]报道,ZrO2层具有优良的阻氢特性,膜层越厚,致密性越好,阻氢效率越高。因此,锆基体及微弧氧化膜试样蒸汽氧化后,锆基体及内层氧化膜中并未明显探测到氢元素。
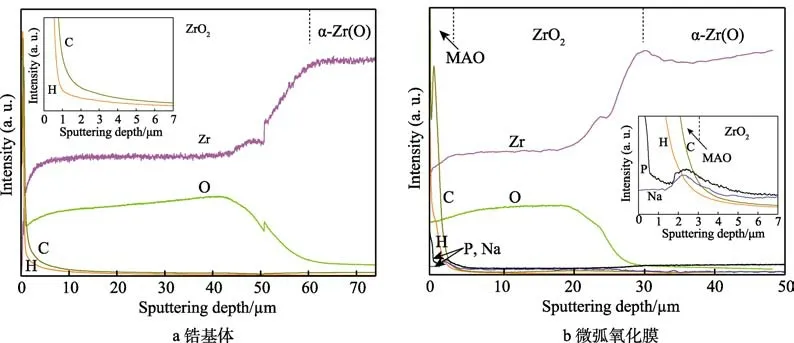
图7 锆及微弧氧化膜在1000 ℃蒸汽中氧化3600 s 后的GDOES 成分深度分布Fig.7 GDOES composition depth profiles of bare and MAO-coated pure Zr after 3600 s steam oxidation at 1000 ℃: (a) bare Zr,(b) MAO-coated Zr
2.4 蒸汽氧化后氧化层相组成分析
图8 为锆基体及微弧氧化膜在1000~1200 ℃蒸汽氧化后的XRD 谱图。图8a 显示,锆基体在1000~1200 ℃蒸汽环境中氧化3600 s 后,氧化层由M-ZrO2和少量的T-ZrO2相组成,这同文献报道结果一致[44]。
图8b 显示,微弧氧化膜试样在1000~1200 ℃蒸汽中氧化后,相组成与锆基体表面氧化层一致。图中还观察到,1000 ℃蒸汽氧化后,微弧氧化膜试样表面氧化层增厚,M-ZrO2相的衍射峰随之增强,α-Zr相的衍射峰消失(见图1d)。随着氧化温度的提高,微弧氧化膜试样中T-ZrO2相的衍射峰随之减弱,表明蒸汽氧化过程中微弧氧化膜中的T-ZrO2相转变成M-ZrO2相[14]。
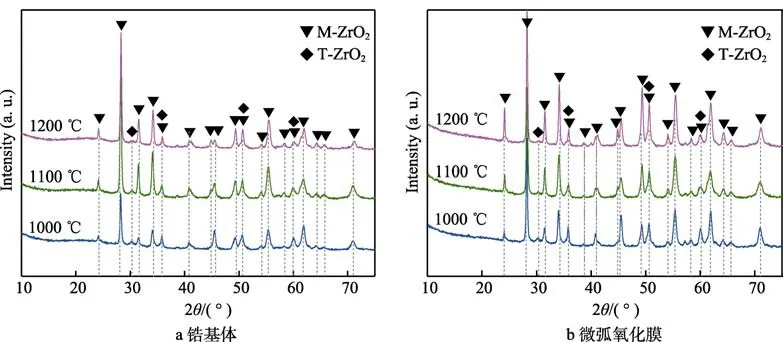
图8 锆及微弧氧化膜不同温度蒸汽氧化后的XRD 谱图Fig.8 XRD patterns of bare and MAO-coated pure Zr after 3600s steam oxidation at different temperatures: (a) bare Zr, (b)MAO-coated Zr
2.5 锆蒸汽氧化机理分析
氧和氢在β-Zr、ZrO2和α-Zr(O)中的扩散系数方程[47-48]见式(4)—(8)。
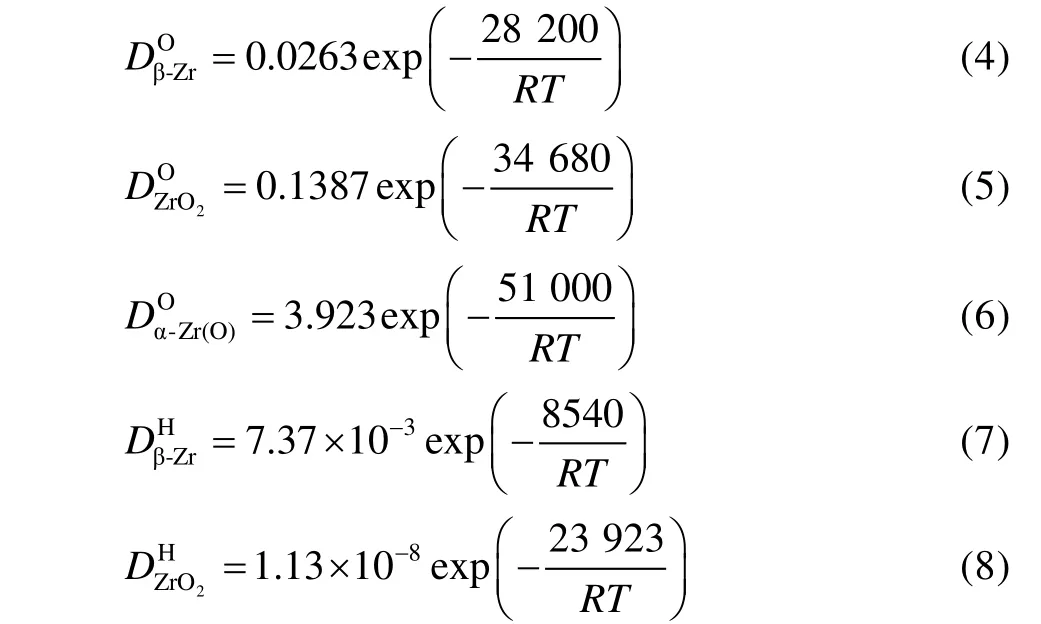
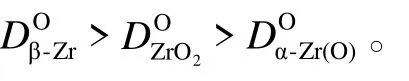
对于微弧氧化膜试样,较厚的α-Zr(O)层形成之前,氧化增重速率取决于氧在微弧氧化膜中的扩散速率。当较厚的α-Zr(O)层形成后,氧化增重速率主要取决于氧在α-Zr(O)层中的扩散速率,这是由于同一温度下Zr(O)比低约1 个数量级。因此,1000 ℃温度下,氧化初期微弧氧化膜抑制了氧向锆基体内部扩散。同时,微弧氧化膜的Kn值低于锆基体,氧化增重也低于锆基体(见图4a)。
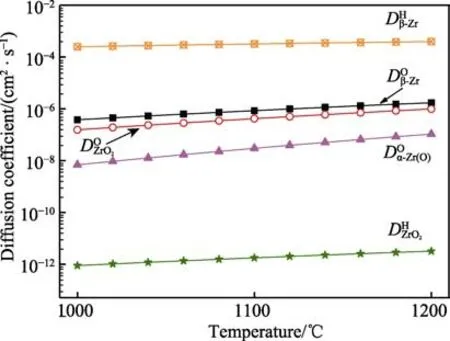
图9 不同温度下氧和氢在β-Zr、ZrO2 和α-Zr(O)中的扩散系数Fig.9 Diffusion coefficients of oxygen and hydrogen in β-Zr,ZrO2 and α-Zr(O) at different temperatures.
另一方面,在1100 ℃和1200 ℃温度下,纯锆表面薄的微弧氧化膜可能因较大的热应力和生长应力而破坏[20]。Lee[52]发现,Zr-1Nb-1Sn-0.1Fe 合金表面预氧化膜经1012 ℃蒸汽氧化后,预氧化膜中出现少量的空洞。随着氧化温度升高至1100 ℃和1200 ℃,预氧化膜中的空洞逐渐生长,并伴随着晶间裂纹形成。因此,在生长应力、内应力及微观缺陷等的共同作用下,微弧氧化膜在1100 ℃以上失效,几乎丧失了抗蒸汽氧化能力(见图4b 和图4c)。研究发现[53],在与文中相同电解液和电参数条件下,Zr-1Nb 合金微弧氧化膜的厚度可以达到8 μm,比纯锆材料的微弧氧化膜厚得多。但前者的抗蒸汽氧化能力明显高于后者,在1000 ℃蒸汽中Zr-1Nb 合金微弧氧化膜的氧化增重只有Zr-1Nb 基体的56%,在1100 ℃以上则失去防护能力。因此,微弧氧化膜厚度对锆基材料的蒸汽氧化增重有明显影响,但锆及锆合金微弧氧化膜都只对1000 ℃以下蒸汽氧化有防护作用,高于1100 ℃则需要同其他技术结合以提高它们的抗蒸汽氧化能力,锆基材料复合表面处理是后续值得研究的方向。
图9 还显示,同一温度下,氢在ZrO2中的扩散系数()比β-Zr 中的扩散系数()约低8个数量级,表明致密的氧化锆膜可有效抑制氢渗透。随着氧化锆膜层的增厚,大幅度降低了锆基体吸氢速率。因此,锆及微弧氧化膜试样在1000 ℃蒸汽氧化后,H 元素仅富集在厚度约3.0 μm 的外部氧化层中(见图7)。
3 结论
1)纯锆在磷酸盐电解液中制备的微弧氧化膜厚度达到2.5 μm,微弧氧化处理使纯锆与水、正十六烷的接触角增大,降低了纯锆的表面能,增强它的憎水性。
2)蒸汽环境中,锆基体及微弧氧化膜的加速氧化动力学转变温度约为600 ℃。
3)微弧氧化膜在1000 ℃以下提高了锆的抗蒸汽氧化性能,但1100 ℃以上时,锆基体和微弧氧化膜的氧化增重曲线几乎重合,微弧氧化膜丧失了保护作用。
4)蒸汽氧化初期氧原子快速扩散至β-Zr 中。当较厚的α-Zr(O)层和ZrO2层形成后,氧化速率主要取决于氧在α-Zr(O)层中的扩散速率。高温蒸汽环境中,氧化锆层有效阻挡了氢扩散进入锆基体。
5)1000~1200 ℃蒸汽氧化后,锆试样表面氧化层由单斜氧化锆(M-ZrO2)相和少量的四方氧化锆(T-ZrO2)相组成。
猜你喜欢
设备管理与维修(2022年21期)2022-12-28
金属热处理(2022年8期)2022-11-17
石材(2022年3期)2022-06-01
物理学报(2022年2期)2022-02-17
原道(2022年2期)2022-02-17
理化检验-化学分册(2020年5期)2020-06-15
表面工程与再制造(2019年1期)2019-05-11
中国核电(2017年1期)2017-05-17
小猕猴智力画刊(2017年4期)2017-05-04
军事文摘·科学少年(2017年1期)2017-04-26
