
原发性纤毛运动障碍患者的临床特征分析
2021-07-03 03:12李倬哲宋元林
中国临床医学 2021年3期
毕 晶, 李倬哲, 周 磊, 宋元林*
1. 复旦大学附属中山医院呼吸与危重症医学科,上海市肺部炎症与损伤重点实验室,上海市呼吸病研究所,上海 200032 2. 复旦大学附属中山医院青浦分院呼吸科,上海 201700
原发性纤毛运动障碍(primary ciliary dyskine-sia,PCD),又称纤毛不动综合征,是一种罕见病,多为常染色体隐性遗传,少数为X染色体连锁相关的双等位基因突变导致的一种遗传病[1]。目前,尚无PCD发病率的准确数据,国外研究[2]报道发病率为1∶40 000至1∶2 000。PCD患者常因反复呼吸道感染、支气管扩张、慢性鼻窦炎、中耳炎、男性不育等表现就诊,另有50%的患者存在内脏转位,称为Kartagener综合征[1,3]。本研究回顾性分析2009年1月至2020年12月复旦大学附属中山医院收治的20例PCD患者的临床特征,并进行相关的文献回顾,以期提高临床医生对该病的认识。
1 资料与方法
1.1 研究对象 回顾性选择2009年1月至2020年12月复旦大学附属中山医院收治的PCD患者20例。纳入标准:(1)符合《原发性纤毛运动障碍诊断与治疗中国专家共识》[4]中关于PCD的诊断标准;(2)Kartagener综合征患者,具有典型的三联征,即支气管扩张、鼻窦炎和内脏反位。
1.2 观察指标 收集20例患者的病例资料,包括(1)人口学资料:性别、年龄、民族。(2)家族史:父母有无近亲结婚,家族成员中有无类似的症状或已确诊PCD的患者。(3)临床症状:记录患者起病时主要症状,出现症状的时间及确诊时间。(4)体征:杵状指、肺部湿啰音或干啰音。(5)胸部CT影像:记录患者胸部支气管扩张病灶部位及类型。鼻窦CT影像: 记录患者鼻窦炎病灶部位。(6)肺功能及血气分析检查结果。(7)痰培养结果。(8)精液检查:男性患者在复旦大学附属中山医院泌尿外科完成精液化验。(9)鼻黏膜或支气管黏膜电镜检查:患者在稳定期(近4周无呼吸道感染)行鼻内窥镜或支气管镜下鼻黏膜或支气管黏膜活检,活检标本予2%戊二醛固定,采用超薄切片并染色后,透射电镜下观察纤毛超微结构有无异常。(10)治疗情况:记录患者治疗药物的种类、剂量及疗程(包括止咳、化痰药物、支气管舒张剂及吸入糖皮质激素等)。记录患者非药物治疗的种类及时间,包括氧疗、无创通气、物理疗法、因咯血行介入治疗或外科手术、肺移植、中耳炎手术、鼻窦炎手术、辅助生殖治疗等。

2 结 果
2.1 人口学特征及临床特征 20例PCD患者中,女性9例,男性11例,均为汉族,发病年龄0~15岁,平均发病年龄(7.25±4.90)岁,确诊年龄8~41岁,平均确诊年龄(25.15±9.80)岁,从发病到确诊平均(17.9±8.77)年。其中3例患者有近亲结婚家族史,5例患者家族成员中有PCD典型临床表现。
自幼反复咳嗽(19/20)、咳痰(19/20)是PCD患者的主要症状。大部分患者合并鼻窦炎症状,如鼻塞、流脓涕(14/20);部分患者合并中耳炎症状,如听力下降、外耳流脓(5/20);其他症状包括咯血(4/20)、活动后喘息(3/20)、不孕不育(2/20)。其中11例患者存在内脏转位,诊断为Kartagener综合征。主要体征包括消瘦(6/20)、杵状指(2/20)、肺部湿啰音(2/20)。
2.2 影像学特征 胸部CT结果(图1A)示20例患者均存在支气管扩张征象,其中17例为弥漫性病变(2个或以上肺段受累),3例为局灶性病变(1个肺段受累)。下叶(17/20)、右中叶(16/20)及左舌段(14/20)为最常见的受累部位。鼻窦CT结果(图1B)示大部分患者(18/20)均有鼻窦炎表现,其中上颌窦炎最为常见(18/18)。
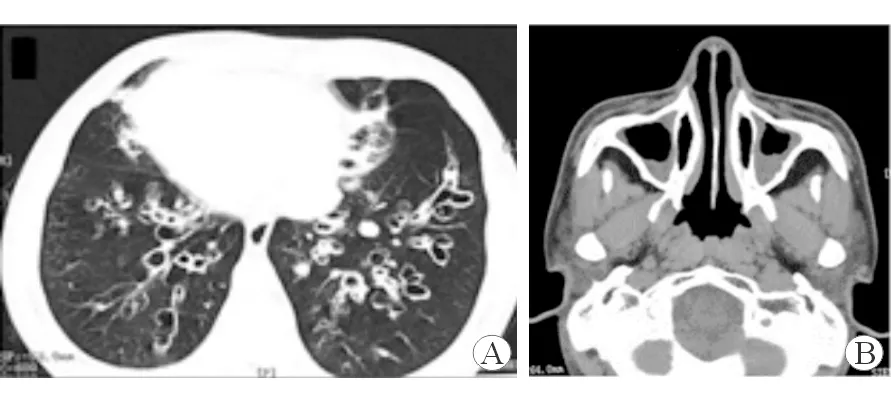
图1 PCD典型病例的影像学资料
2.3 肺功能及血气分析 20例PCD患者中共有14例完成了肺功能检查。其中阻塞性通气功能障碍8例,限制性通气功能障碍伴有弥散功能障碍2例,混合性通气功能障碍3例,患者肺功能完全正常1例。患者1秒钟用力呼气容积(forced expiratory volume in 1 second, FEV1)占预计值%为(55±23)%;肺活量(forced vital capacity, FVC)占预计值%为(70±25)%;FEV1/FVC平均为(56±19)%;肺总量(total lung volume, TLC)占预计值%平均为(81±18)%。14例患者有静息时动脉血气结果,大部分患者(12/14)均存在不同程度的低氧血症,1例患者存在呼吸衰竭。血氧分压(PaO2)平均为(73±6) mmHg (1 mmHg=0.133 kPa)。
2.4 痰微生物学特征 共获得16份合格的痰培养标本,仅6例患者的痰培养阳性,其中铜绿假单胞菌3例、流感嗜血杆菌2例、金黄色葡萄球菌1例。结核分枝杆菌培养均为阴性。
2.5 精子活性检测 共有5例男性患者接受精液分析,5例患者均存在不同程度的精子活动力下降,精子活动率平均为(14±2)%。
2.6 鼻黏膜或支气管黏膜电镜检查 共有9例患者完善了鼻黏膜或支气管黏膜的电镜检查。正常纤毛电镜下超微结构(图2A)表现为横截面呈“9+2”结构,即由9对外周微管环绕1对中央微管组成,连接于外周微管向内、外侧伸展的分别是内动力臂(inner dynein arms,IDA)、外动力臂(outer dynein arms,ODA),IDA通过辐射臂与中央微管间相互连接固定。而透射电镜结果(图2B)示:9例患者中5例存在ODA缺失,其余纤毛结构异常包括ODA和IDA联合缺失2例,IDA缺失合并微管结构紊乱1例,中央微管缺失1例。

图2 正常纤毛超微结构示意图和PCD典型病例透射电镜观察图
2.7 治疗情况 14例患者长期口服止咳、化痰药物,如乙酰半胱氨酸、桉柠蒎肠溶软胶囊。 8例存在阻塞性通气功能障碍的患者应用长效β-受体激动剂治疗。1例患者需长期家庭氧疗及间断无创呼吸机辅助通气。2例患者因中耳炎行鼓膜切开术,1例患者因中耳炎致听力下降行鼓膜成形术。2例患者因不孕不育行辅助生殖技术。
3 讨 论
PCD属常染色体隐性或X连锁相关的双等位基因突变的遗传疾病,其中约50%病例合并内脏转位,称为Kartagener综合征。本病早在1904年就由Sievert首次报道,但直到1933年,瑞士医生Kartagener报道了4例鼻窦炎、支气管扩张合并右位心的病例,并首次将这3种疾病联系在一起,将其命名为Kartagener综合征。基因突变导致纤毛结构和(或)功能异常,各器官的纤毛运动异常,导致了PCD患者的各种临床症状和体征[1-2]。
PCD的临床表现随发病年龄、有无脏器转位、临床症状的严重程度存在差异,尤其是不伴有内脏转位的PCD患者,其临床表现缺乏特异性,易被误诊或漏诊。PCD患者常自幼反复发生呼吸道感染,可伴有鼻息肉、鼻窦炎或副鼻窦发育不全等。患者中耳和耳咽管纤毛功能异常,可伴慢性复发性中耳炎、鼓膜穿孔、流脓。男性患者精子失去摆动能力可致不育症。女性患者输卵管纤毛功能异常可致生育力下降[1-2]。目前,国内报道的PCD患者男性更多见,均为幼年发病,诊断年龄为4个月至74岁[5]。常见的临床表现包括反复咳嗽、咳痰、鼻窦炎、分泌性中耳炎、不孕和不育等,合并内脏反位的高达95.5%。而国外报道仅40%~55%的PCD患者存在内脏转位[1],提示国内不合并内脏转位的PCD患者漏诊率极高。本研究确诊的20例PCD患者从发病到确诊时间长达近20年,尤其是不合并内脏转位的PCD患者极易在呼吸科或五官科被误诊为支气管扩张、慢性鼻窦炎。如何进一步提高我国广大呼吸内科医师对PCD的认识,尤其是早期诊断不合并内脏转位的PCD患者是目前亟待解决的难点。本研究团队较早在国内展开遗传性支气管扩张,尤其是PCD的相关研究,自2010年在Chest发表PCD病例报告[3]起,逐步建立PCD患者随访队列,围绕PCD的临床表现、遗传学特征、诊断及治疗开展临床研究,并于2020年牵头完成《原发性纤毛运动障碍诊断与治疗中国专家共识》[4]。
国内外关于PCD的指南和专家共识[1,4,6-7]均推荐对具有以下至少2项PCD典型临床特征的患者进行PCD筛查试验,包括自幼持续咳嗽咳痰、自幼持续性鼻塞、足月儿发生不明原因的新生儿呼吸窘迫综合征、内脏异位。此外,由于PCD是遗传学疾病,临床还应该重视对患者家族史的询问。本研究确诊的20例PCD患者中3例患者有近亲结婚家族史,5例患者家族成员中有PCD典型临床表现的患者,其中有2例患者为同胞兄弟。因此,对PCD患者的同胞兄妹,特别是有提示PCD症状者也应进行PCD的筛查。
既往曾用糖精试验筛查疑似的PCD患者,鉴于其灵敏度和特异度均较低,已被国外PCD的诊断指南[1,6]摒弃。目前欧洲呼吸学会(European Respiratory Society, ERS)和美国胸科学会(The American Thoracic Society,ATS)关于PCD的最新诊断指南均推荐鼻呼出气一氧化氮(nasal nitric oxide, nNO)测定作为PCD的筛查试验。许多研究[1,6]表明PCD患者nNO显著降低,且该项检查为非侵入性,操作简便,灵敏度和特异度均超过90%。鉴于国内目前鲜有开展nNO测定的医疗机构且缺乏标准的操作规范,未来亟需大力推广nNO的规范化检测,以用于PCD的筛查。然而,仅凭nNO降低并不能诊断PCD,其他窦-肺疾病如囊性纤维化、慢性鼻窦炎以及成年人弥漫性细支气管炎患者nNO也会降低。此外,有研究[8]报道,一部分PCD患者的nNO值正常。未来尚需通过大样本的PCD队列研究来建立中国人群PCD患者的nNO诊断阈值。
目前,国外对PCD患者纤毛形态及功能检测的主要方法有透射电镜、高速视频成像分析和免疫荧光分析法[1,6]。高速视频成像分析和免疫荧光分析法均需要大量的专业技术知识和设备,且成功率较低,不推荐作为PCD的确诊试验。目前国内仍以电镜检查作为确诊PCD的主要手段,PCD患者最常见的显微结构异常是ODA缺失(26%~59%)及ODA和IDA联合缺失(6%~39%)[1],而本研究中的PCD患者中最常见的是ODA缺失,与文献报道相符。国内外PCD的诊断指南及专家共识均推荐透射电镜观察纤毛超微结构作为PCD的诊断试验。值得注意的是,国外研究[6]报道,多达30%的PCD患者可以在电镜下观察到正常的纤毛超微结构。因此,对于高度怀疑PCD的患者,即使电镜检查正常仍不能排除PCD,可进一步行基因检测来协助诊断。
至今已证实的与PCD相关的双等位突变基因有40余个[9-10],近十年来国内共报道20余例基因突变的PCD病例[11-13]。本中心近期对7例Kartagener综合征患者的外周血进行全外显子测序,发现4个异常的DNA AF1突变位点(NM_178452.6:c.3G>A、c.124+1G>C、c.509delG和c.943A>T)[14]。欧美国家已将基因检测作为PCD的诊断试验[1,6],国内受限于高昂的检测费用,缺乏专业的检测结构及分析人员,因此在国内疑似PCD的患者中广泛开展基因检测以发现中国PCD人群的遗传学特征仍任重而道远。
作为一种遗传性疾病,迄今为止尚无针对PCD纤毛功能障碍的特异性治疗药物。目前推荐的治疗方案类似其他病因引起的支气管扩张的治疗,缓解期以化痰药物增加引流,以及增强抵抗力为主,对反复发生肺部感染者,应加强物理或药物祛痰治疗,长疗程抗生素治疗方案是否能使PCD患者获益,目前还没有足够的循证医学证据。建议肺功能显示阻塞性通气功能障碍者吸入长效支气管舒张剂[1,4,6]。目前PCD的治疗仍以对症治疗为主。随着遗传学及分子生物学的不断进展,已有研究可成功模拟PCD的动物模型,或可用于PCD基因治疗研究。
综上所述,国内目前对于不合并内脏转位的PCD患者漏诊率仍较高,亟需提高广大基层医院呼吸内科医师对PCD的认识,早期识别疑似PCD的患者,并尽快转诊至有PCD诊断能力的区域医疗中心。未来需开展全国多中心PCD诊断和治疗临床研究,为国内PCD患者的诊治提供更多的循证医学证据。
猜你喜欢
中国临床医学影像杂志(2022年2期)2022-05-25
医学研究生学报(2021年4期)2021-12-02
辐射研究与辐射工艺学报(2021年2期)2021-05-06
临床与实验病理学杂志(2021年3期)2021-04-25
实用皮肤病学杂志(2020年2期)2020-05-11
散文诗世界(2019年6期)2019-09-10
意林·全彩Color(2019年7期)2019-08-13
家庭科学·新健康(2018年11期)2018-11-16
中国医学创新(2018年28期)2018-02-23
云南中医中药杂志(2018年10期)2018-01-19
