
Cobalt anchored CN sheet boosts the performance of electrochemical CO oxidation∗
2021-06-26 03:05:00XuLiu刘旭JunChaoHuang黄俊超andXiangMeiDuan段香梅
Chinese Physics B 2021年6期
关键词:刘旭
Xu Liu(刘旭), Jun-Chao Huang(黄俊超), and Xiang-Mei Duan(段香梅)
School of Physical Science and Technology,Ningbo University,Ningbo 315211,China
Keywords: first-principles calculations,single-atom catalyst,CO oxidation,rate-limiting reaction barrier
1. Introduction
As greenhouse gases like CO get increased,[1]researchers are trying their best to find solutions to decrease the content of CO in the air.[2]One of the most promising methods is to convert CO to CO2by a highly effective catalyst.[3,4]So far,considerable efforts have been made to develop novel and efficient catalysts for CO oxidation. Some typical noble metals can effectively catalyze CO oxidation, such as Pt,[5]Pd,[6]Ru,[7]and Au.[8]However, considering the low cost, environmental friendliness, and outstanding thermal stability, researchers start to develop noble-free metals gradually and achieve certain success in CO oxidation.[9]Adhering to further increase the catalytic performance and reduce the cost,the view of researchers turns to single-atom catalysts (SACs)[10]for their high site density,high specify,and low cost.[11]
Two-dimensional (2D) materials are extensively applied in various catalytic reactions as substrates owing to their large surface area, high thermal stability, and easily manufacture, such as graphene,[12,13]MoS,[14,15]hexagonal boron nitride monolayers (h-BN),[16,17]and graphic carbon nitride(CN,[18–20]C2N,[21,22]and C3N4[23–25]). Many 2D materials are rich in electron pairs to combine with metal ions, and it seems easy to recognize active sites. Besides,due to the crude or artificial vacancies, 2D materials can be used as prominent supports to host metal atoms and get high stable catalyst. In the experiment, SACs can be synthesized using the mass-selected soft-landing technique,improved wet chemistry methods, or atomic layer deposition methods. Many studies showed that defects of the support materials could serve as anchoring sites for metal clusters and even single atom.[26]
It is reported that carbon nitride (CN), successfully synthesized in the experiment, can be applied in He separation,[25]H2storage,[27]water decomposition,[28,29]and CO oxidation.[20]Co atom, as an ideal noble-free metal, has a good performance in CO oxidation, even surpassing traditional noble metal catalysts. For instance,experimental works showed that the Co atom in Co3O4shows strong catalytic activity in CO oxidation at low temperature.[30,31]However,the reaction barrier of Co-anchored graphene(Co@Gra)is as high as 0.65 eV,[32]which cannot be carried out at room temperature. In order to improve the performance, researchers start to introduce defects in graphene-based SAC,and the reaction barrier of Co@Sv-Gra is decreased by 0.23 eV.[32]Co implanted h-BN and g-C3N4are also reported as excellent catalysts for CO oxidation, and the reaction barriers of Co@h-BN[33]and Co@g-C3N4[34]are 0.41 eV and 0.21 eV,respectively. The catalytic behavior of Co-anchored CN in the CO oxidation reaction is worth studying.
Based on first-principles calculations,we investigated CO oxidation on Co anchored planar CN sheet. Our results show that the Co atom(s) can perfectly bind to the CN monolayer and maintain the stability of the whole system. When O2and CO molecules are adsorbed over Co/2Co@CN, the gas molecules are activated through electron “acceptance donation” interaction between gas molecules and the transition metal. Furthermore, Co anchored CN systems possess superior catalytic activity toward the CO oxidation reaction.
2. Computation details
The calculations are performed based on density functional theory (DFT) by the Viennaab initiosimulation package (VASP).[35]The Perdew–Burke–Ernzerh (PBE) functional of generalized gradient approximation (GGA) with the semi-empirical van der Waals (vdW) method of Grimme(DFT-D2) was used to handle the exchange and correlation functionals.[36,37]The cutoff energy is set to be 500 eV.The convergence tolerance for the geometry optimization is 10−4eV for the energy and 10−2eV/˚A for the force. A vacuum space of 20 ˚A is adopted to avoid interaction between two adjacent images. The reciprocal space is sampled with a 3×3×1k-point grid generated automatically using the Monkhorst-Pack method[38]for optimization. A HubbardUterm was added to the PBE functional (DFT+U) to describe partially filled d orbitals. The on-site Coulomb interaction(U)of 4 eV and exchange interaction(J)of 1 eV were applied.[39]Additionally,we used the climbing image nudged elastic band(CI-NEB) method[40]to search for transition states to investigate the reaction paths. Bader charge analysis is adopted to obtain the number of transferred electrons.[41]The first principles molecular dynamics(MD)simulations at 300 K with a time step of 1 fs are performed to check the structural stability.
Taking Co@CN as an example, the binding energy (Eb)of Co on CN is defined as the energy difference between the Co atomically deposited CN and the separated CN plus the freestanding Co atom,that isEb=ECo@CN−ECN−ECo. The more negative binding energy,the more energy advantage the anchoring of Co on the substrate.
The adsorption energy (Ea) of the gas molecules is obtained by the energy difference between the absorbed Co@CN and the gaseous species plus the bare Co@CN.Eacan be expressed asEa=Eadsorbate+Co@CN−Eadsorbate−ECo@CN. The charge density difference ∆ρis used to describe the electronic interaction between the substrate and the adsorbate, ∆ρ=ρadsorbate/support−ρadsorbate−ρsupport. Hereρadsorbate/support,ρadsorbate, andρsupportare the corresponding charge densities of the combined system,the free adsorbate,and the bare support,respectively.
3. Results and discussion
3.1. One and two Co atoms anchored CNs
The calculated lattice constant of CN is 7.13 ˚A, the C–N and C–C bond lengths are 1.35 ˚A and 1.52 ˚A,respectively.The results are in good agreement with the reported values of 7.12 ˚A, 1.34 ˚A, and 1.54 ˚A.[27,28]A 2×2×1 supercell containing 24 carbon and 24 nitrogen atoms was established. The intrinsic hole surrounded by six sp2bonded nitrogen atoms is beneficial for Co atom anchoring. We consider three possible sites for Co landing on CN,find the most favorable configuration is the one shown in Fig. 1(a), where the Co atom bonds to two edge N atoms with a bond length of 1.85 ˚A and keeps
its planar geometric structure undistorted(see Fig.1(c)). The most stable geometry for two Co atoms on CN is shown in Fig.S1(a),with the Co–N bond length of 1.81 ˚A and the Co–Co distance of 2.39 ˚A. The corresponding binding energy is calculated to be−3.77 eV and−3.26 eV/atom for Co@CN and 2Co@CN, respectively, indicating the strong interaction between the Co atom and CN monolayer. For comparison,the data are listed in Table 1, where the cohesive energies per atom of the bulk metalsEc(−4.39 eV) are taken from experiments.[42]
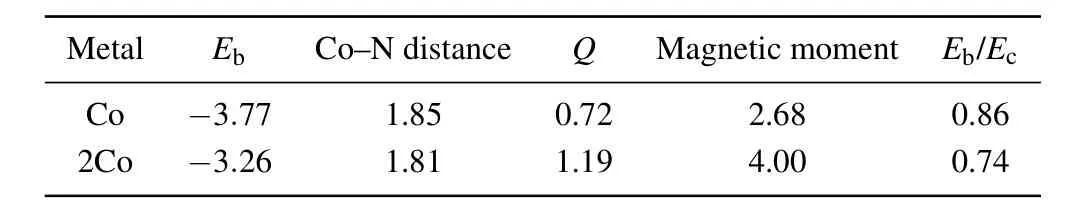
Table 1. The binding energies (eV) for Co, Bader charge Q (|e|) from Co to the substrate,the bond length of Co−N(A˚),magnetic moment(µB),and the ratio of Eb/Ec for the most favorable Co adsorptions on CN.
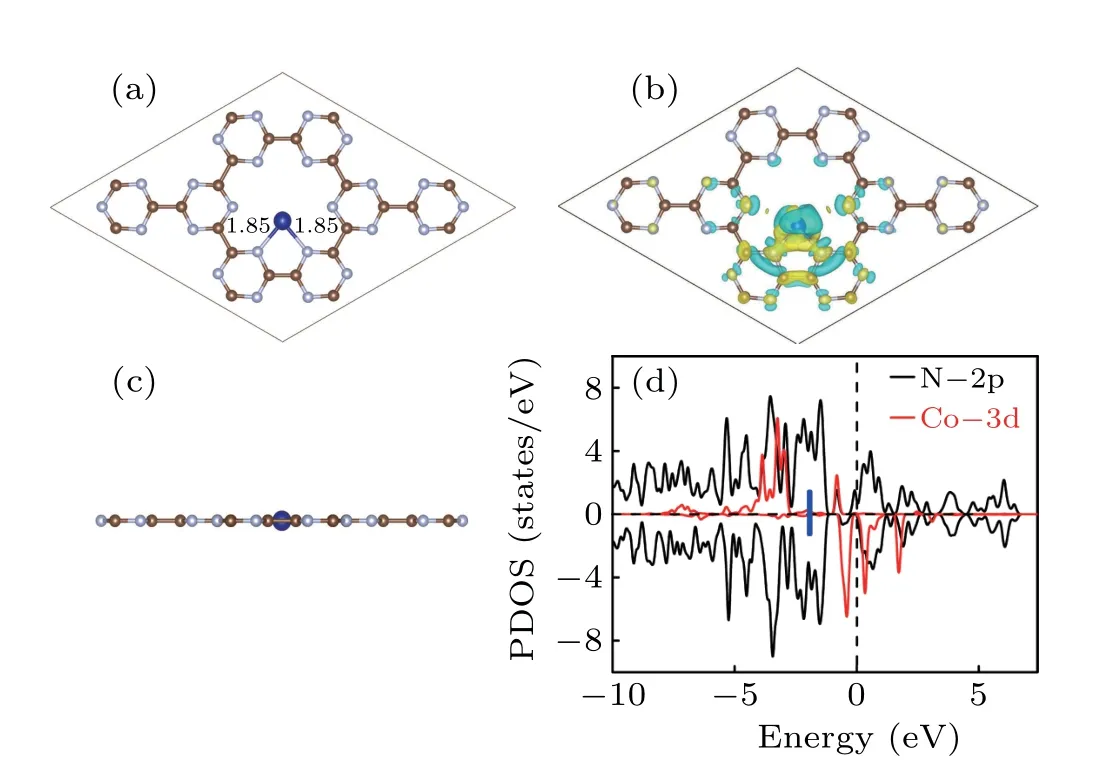
Fig. 1. The top (a) and side view (c) of optimized atomic configuration, the charge density difference plots (b) with an iso-surface value of 0.003|e|/Bohr3,as well as spin-polarized PDOS(d)for Co@CN.The charge accumulation and depletion regions are represented in yellow and blue, respectively. The bold blue short line points to the d-band center (−1.77 eV)of Co atom.
The transition metals have a tendency of forming clusters,then we investigate the stability of Co@CN by CI–NEB and MD methods. As shown in Figs. 2(a) and 2(b), the diffusion barrier for Co from one stable site to another in the neighboring hole is 2.62 eV.Such a large diffusion barrier conforms that the single Co atom binds with the CN monolayer steadily and hard to move. On the other hand,the largeEb/Ecratios of 0.86 and 0.74 for single and double cobalt on the substrate manifest that Co atoms prefer a 2D growth morphology on CN.Furthermore, MD results manifest that the structure remains undistorted at room temperature (see Figs. 2(d) and 2(e)). Charge transfers from Co and 2Co to CN monolayer are 0.72|e| and 1.19|e|,respectively. Comparing the energy band structure of pure CN monolayer with that of Co@CN and 2Co@CN in Fig.S2,the bands dominated by N-2p orbitals in Co@CN and 2Co@CN show that there is a semiconductor to metal transition.
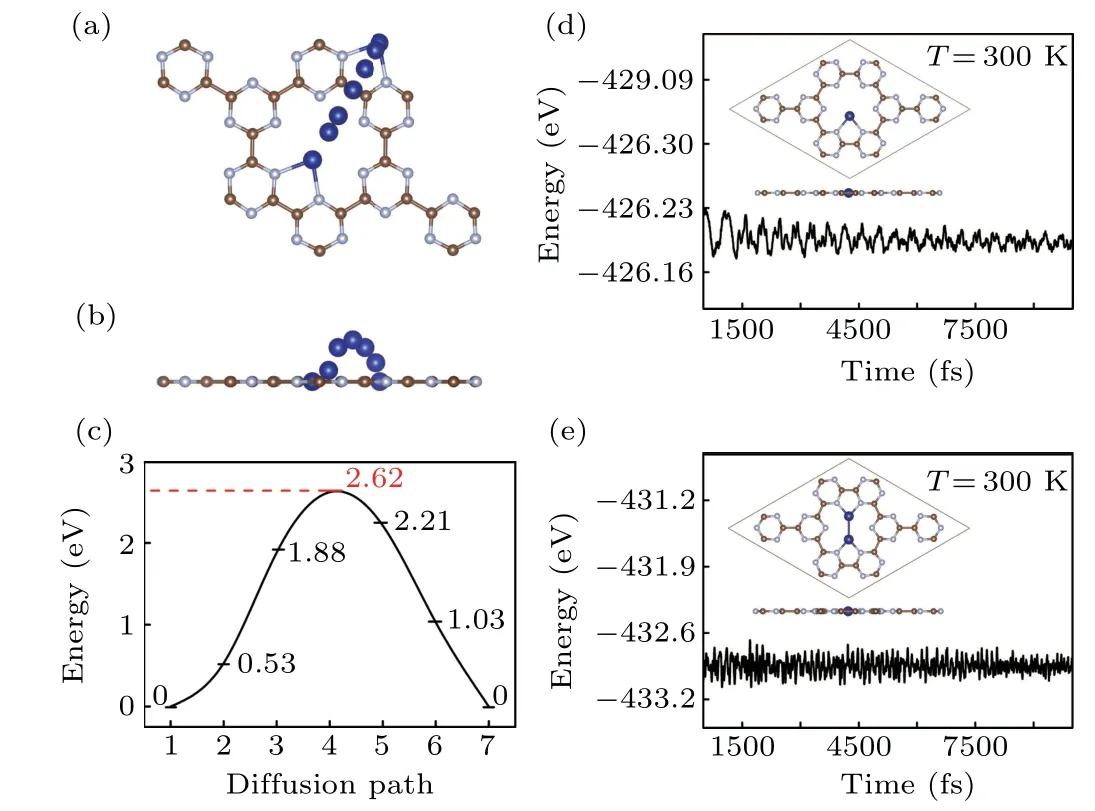
Fig.2. The top(a)and side view(b)of diffusion path diagram and relative energy(c). The results of molecular dynamics(MD)simulations(d)and(e).
As shown in Fig. 1(b) and Fig. S1(b), the charge rearrangement mainly occurs among the Co atom(s) and the twoedged N atoms. To better understand its physical mechanism, partial density of states(PDOS)is plotted in Fig.1(d)and Fig. S1(d). The overlapped peaks caused by orbital hybridization appearing between Co-3d and N-2p states illustrate that the single Co atom(s)can be viewed as an active site. The insertion of the second Co atom proves the ability of catalytic because the d-band center deviates from the Fermi level.[43]The magnetic momentum for Co and 2Co@CN is 2.68µBand 4.0µB,respectively.
3.2. Adsorption of species involved in the CO oxidation
The thermodynamics of a specific reaction are strongly affected by the stability of reactants, intermediates, transition orbitals,and products.[44]The ability to bind the gas molecules on catalytic active centers confirms the reaction efficiency.We then investigate the adsorption of reactants CO and O2over Co atom(s)anchored CN.
O2molecule is one of the most important participants in the whole process of CO oxidation. We consider both end-on and side-on adsorption of O2on both Co@CN and 2Co@CN,and find that the adsorption energy via side-on is more negative (see Table 2 and Table S1); therefore, we focus on this configuration. As shown in Fig.3(a),the O2molecule prefers to lie parallel to the plane of the substrate, on top of the implanted Co atom. The two nearest Co–O distances are both 1.8 ˚A, the O–O bond length is 1.34 ˚A, stretched by 0.11 ˚A compared with the value of free O2(g), which indicates that the “donation–acceptance” between Co and edged N in CN that activates the reaction activity of the single Co atom. The charge transfer of 0.58|e|is mainly caused by the reduction of the Co atom and accumulation of O2molecule(see Fig.3(b)).Evidenced by DOS in Fig. 3(c), after adsorption, the Co-3d states are overlapped with O2-2π∗state, the DOS peaks of O2-5σand O2-1πstates are downshifted, indicating that O2molecule accepts electrons from Co-3d orbital.The strong hybridization among them benefits the O2molecule to combine with Co@CN.
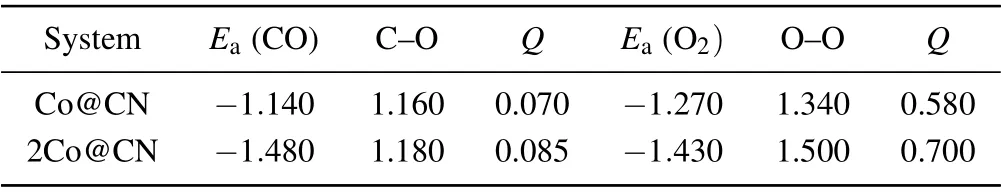
Table 2. The adsorption energies(eV/atom)for CO and O2(side-on),Bader Charge Q(|e|/atom),and bond length(˚A)for the Co implanted CN.
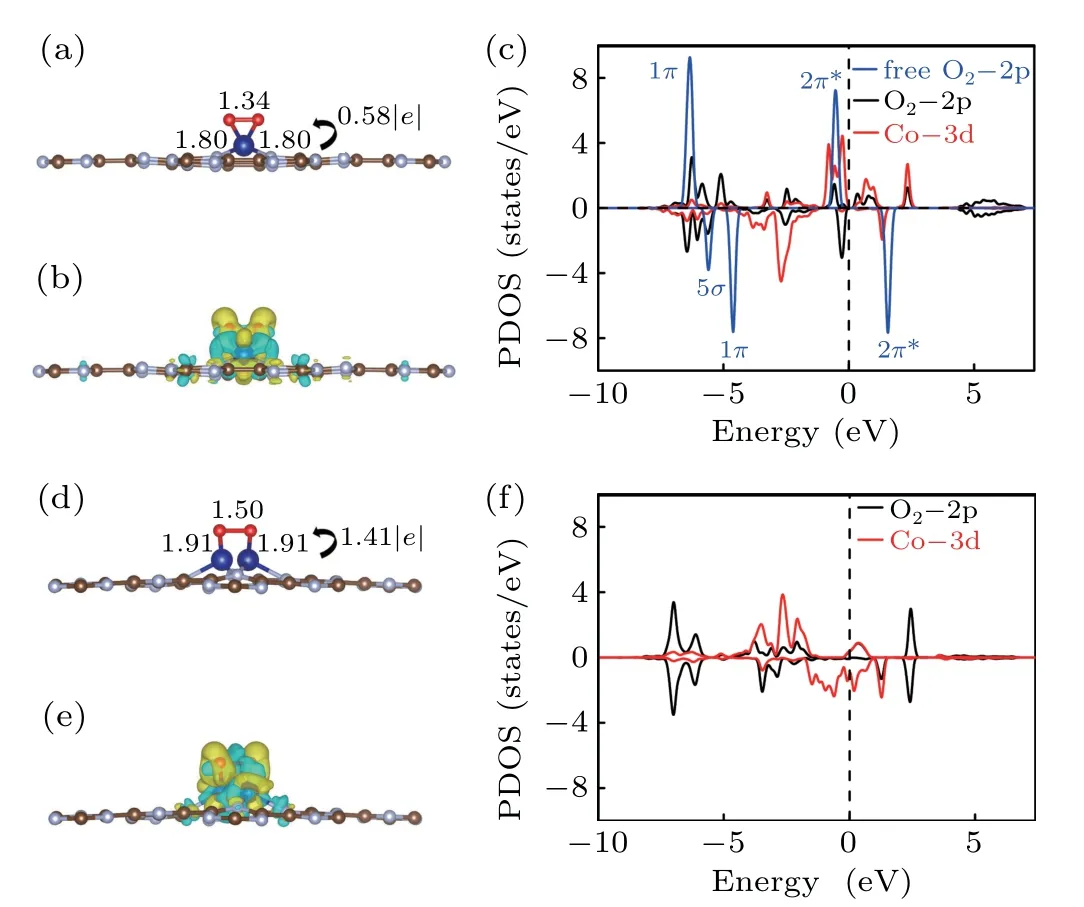
Fig. 3. The side views of the most plausible adsorption structures, the corresponding charge density difference,which iso-surface value is set to 0.003 e/Bohr3 and the PDOS for O2 on Co@CN (a)–(c) and 2Co@CN(d)–(f).
For 2Co@CN shown in Fig. 3(d), the O2molecule adsorbs over the catalyst,like a bridge connecting one Co atom to the other. TheEaof the O2molecule is−1.43 eV/atom,and it is relatively larger than that of the O2molecule on 2Cu@C2N(−0.67 eV/atom). The bond length of O–O(1.50 ˚A)is longer than that of O2over Co@CN. Moreover, Bader charge analysis shows that each Co atom transfers 0.70|e| to the O2molecule. After O2adsorbing, there are several peaks overlapping with Co-3d and O2-2p states at the energy region of−2 eV to−5 eV,as shown in Figs.3(c)and 3(f).For Co@CN,the O2molecule donates 1πand 5σstate electrons to Co-3d state, and the occupied Co-3d states feedback electrons to 2π∗state of O2molecule, which is evidenced by the spindown peak near Fermi level. The ability to adsorb O2of both Co@CN and 2Co@CN is strong, especially the latter, with more charge(0.70|e|vs.0.58|e|)transferred from the substrate to O2molecule,the length of O–O bond is more elongated.
CO interacts strongly with the Co@CN.For the most stable configuration,the calculatedEaof CO is−1.14 eV,which is close to the value of O2(−1.27 eV),so CO poisoning may be avoided. As presented in Fig. 4(a), the CO molecule is not perpendicular to the Co@CN plane but is inclined. The length of the C–Co bond is 1.82 ˚A. The C–O bond length increases by 0.02 ˚A compared to the value of free CO(g)(1.14 ˚A). The charge transfer is 0.07|e| from the substrate to CO.From the density of states in Fig.4(c),it can be seen that there is slight charge transfer between CO and the substrate due to the donation of CO-5σstate electrons to Co-3d state and the back-donation of Co-3d state electrons to the CO-2π∗state. For CO on 2Co@CN, the adsorbate stands above the two Co atoms and is almost perpendicular to the CN sheet(see Fig.4(d)). TheEaof−1.48 eV/atom is closer to the value of O2(−1.43 eV/atom);therefore,CO poisoning will not occur.The bond lengths of C–O(1.18 ˚A)are also longer than that of the CO molecule on Co@CN.Moreover,Bader charge analysis shows that the adsorbed CO molecule gains 0.085|e|/atom from 2Co@CN.The strong interaction between Co and CO is evidenced by the DOS in Fig.4(f). The slight charge transferring is mainly caused by the donation of CO-4π∗,1π,5σelectrons to Co-3d state and back donation of Co-3d electrons to CO-2π∗. Like the adsorption of O2,the more electrons transfer from the substrate to the CO molecule,the larger the C–O bond length.
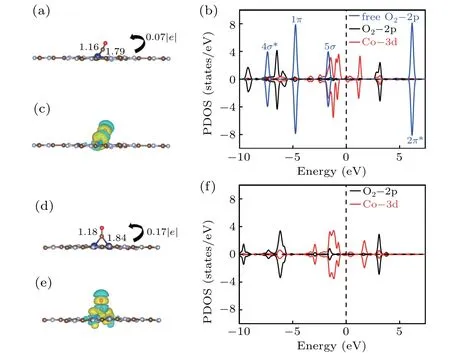
Fig. 4. The side views of the most stable adsorption structures, the corresponding charge density difference and the PDOS for CO on Co@CN(a)–(c)and 2Co@CN(d)–(f).
We also consider the final products O atomic and CO2adsorption on both catalysts and summarize the data in Table S1. TheEaof O atom absorbed on Co@CN and 2Co@CN is−3.94 eV and−3.14 eV, respectively. Due to the strong hybridization between the O-2p and Co-3d states, the Co–O bond length is all less than 2 ˚A. The bond length of the two C–O bonds,as well as the angle of O–C–O,is the same as that of free CO2(g). TheEaof CO2is−0.42 eV and−0.34 eV,respectively,both are less than 0.50 eV,indicating that CO2is easy to desorb.[33]
3.3. CO oxidation on Co@CN and 2Co@CN
Generally, the CO oxidation on catalysts can mainly be performed by three typical reaction mechanisms, namely Eley–Rideal (ER), Langmuir–Hinshelwood (LH), and termolecular Eley–Rideal (TER) mechanism. Considering the recycling of catalysts,we take catalysts themselves as the initial state in three mechanisms.
The ER,a mechanism of single active state participating in the reaction,is initiated by the direct reaction of gaseous CO molecules with the adsorbed O atom at the reaction centers resulting in the activation of O2to form a carbonate-like CO3intermediate or a final product of CO2. The dissociation of O2is the rate-limiting step. For Co@CN,the interaction with CO is as strong as that with O2,which can avoid CO poisoning. The configuration of physically adsorbed CO above the pre-adsorbed O2is selected as the initial state(IS2). As shown in Fig. S3, a chemically adsorbed atomic O and a physically adsorbed CO2molecule are considered as the final state(FS).In IS2, when the CO molecule approaches the activated O2,the O–O distance increases from 1.34 ˚A to 1.35 ˚A.The insertion of CO is exothermic; the breaking of the O–O bond and the formation of the new C–O bonds need to cross a barrier of 0.73 eV.Then it forms carbonate–like CO3(TS1). The CO3in TS1 dissociates by scission of one C–O bond attached to the Co atom. The process has an energy barrier of 3.14 eV(from MS1 to TS2). As a result, there leaves a physically adsorbed CO2and an O atom adsorbing on the Co@CN(FS).CO2adsorption on Co@CN is quite weak,and entire CO oxidation is exothermic at 300 K.The ER mechanism is unfavorable due to its large reaction barriers(>3 eV).The reaction on 2Co@CN is shown in Fig.S5. The entire reaction progress of 2Co@CN is similar to that of Co@CN,and its reaction barrier is 2.65 eV.
The LH,a mechanism of double active states(CO+O2)participating in the reaction, starts with the interaction between the co-adsorbed CO and O2molecules for forming a peroxide-like OCOO intermediate and then the O–O bond breaks. And there leaves a physically adsorbed CO2together with an adsorbed atomic O like the final state of ER.As shown in Fig.S4,the co-adsorption of CO and O2on Co@CN is selected as the initial state(IS).Then crossing an energy barrier of 0.25 eV,CO and O2are parallel with end-on configurations(TS1). The CO and O2are activated,the O–O and C–O bonds are elongated. The peroxide-like OCOO has formed (MS1)with new O–O bond(1.63 ˚A).With the scission of the new O–O bond and Co–C in MS,a physically adsorbed CO2is formed together with an adsorbed atomic O(TS2). The reaction from MS1 to TS2 climbs the energy barriers of 0.79 eV.The entire progress is exothermic with a small reaction barrier of 1.69 eV.The reaction could process readily with the barrier of 0.8 eV or less.[44]The findings indicate that CO oxidation on Co@CN is not particularly desirable. Whereas, O2molecule prefers adsorbing on 2Co@CN (IS1). With CO molecule participating in, the O–O bond stretches. Then one of the O atoms approaches to adsorbed CO molecule, and it forms OCOO(MS1). Then it gets dispersed,the corresponding reaction barrier is only 0.62 eV. We conclude that 2Co@CN is superior in terms of a minor reaction barrier. Following this,as shown in Fig.S6,the left adsorbed atomic O will react with another gaseous CO molecule(IS)and form the second physically adsorbed CO2(FS). For Co@CN (2Co@CN), the formation of the second CO2needs to pass over a reaction barrier of 0.28 eV(0.34 eV) and release the heat of 3.82 eV (2.65 eV), respectively.
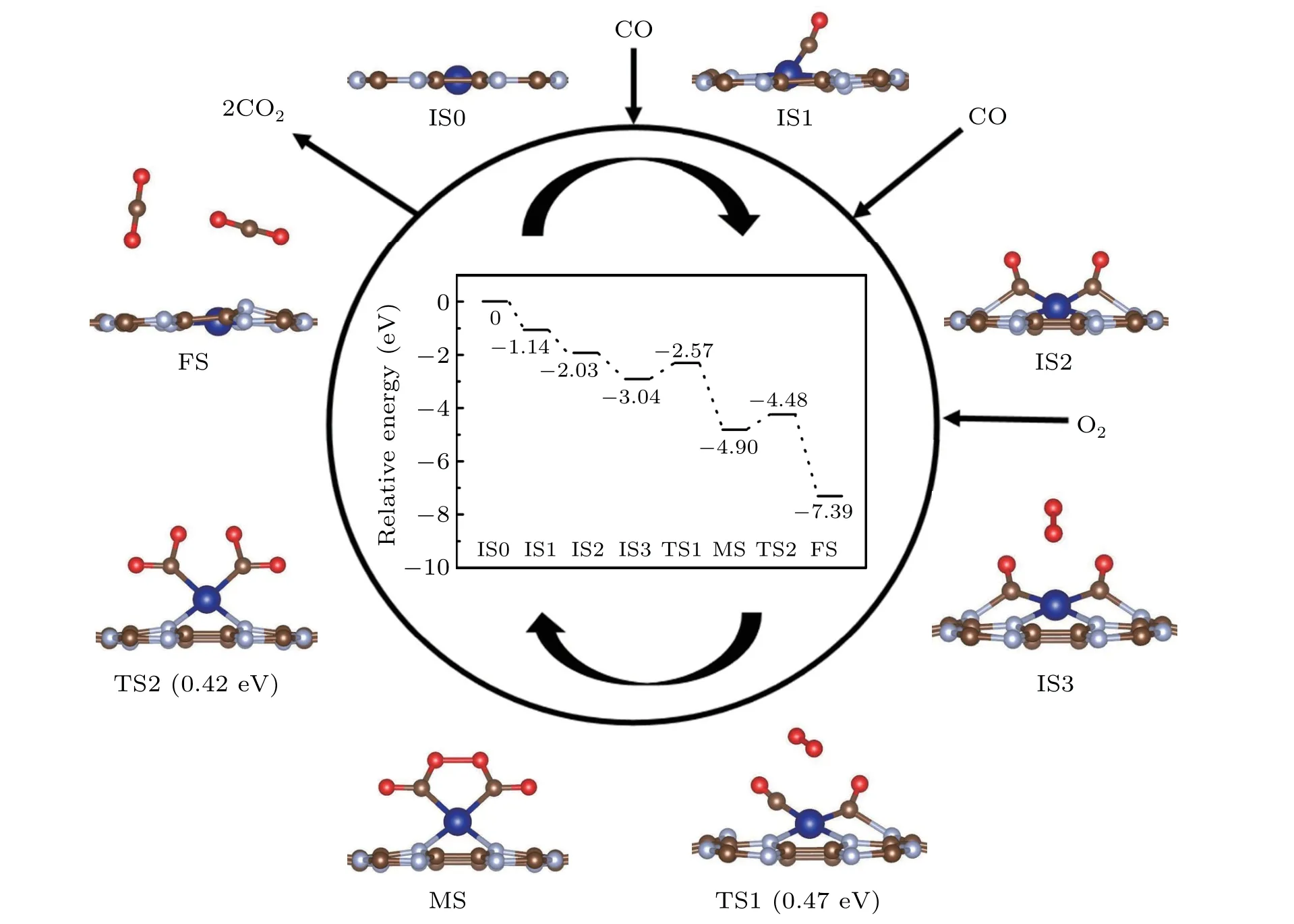
Fig.5. Schematic energy profile corresponding to the configurations(side-view)for CO oxidation on Co@CN through TER mechanism. All energies are given with respect to the reference energies of IS0.
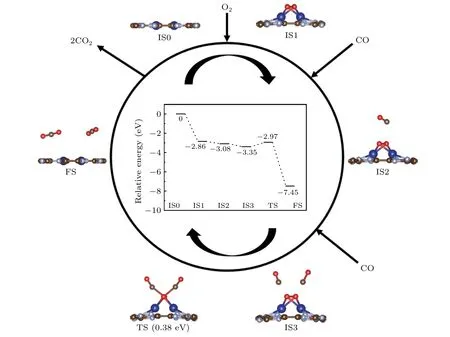
Fig.6. Schematic energy profile corresponding to the configurations(side-view)for CO oxidation on 2Co@CN through TER mechanism.
Similar to the LH mechanism,the TER is a mechanism of double active states (CO+CO) participating in the reaction.Free O2molecule can be activated by the two co–adsorbed CO molecules to form an OCO–M–OCO intermediate. Then CO2molecules gradually stay away from the catalysts after the breaking of the C–Co bond. As shown in Fig.5, two CO molecules are chemically co–adsorbed on the Co site,and one O2molecule approaches them from the top(IS).Then the insertion of O2actives the pre-adsorbed CO with the elongated O–O bond length of 1.28 ˚A (TS1) via adsorbing energy of 0.47 eV.Once a free O2is close enough,two O atoms bind to the C atoms and form an OCO–Co–OCO intermediate(MS).From MS to TS1, as the O–O bond length continues to increase, it finally breaks, and OCO–Co–OCO dissociates into two CO2molecules with a small reaction barrier of 0.42 eV and a huge exothermic energy of 4.35 eV (TS2). Due to the smallEa(−0.42 eV)of CO2on Co@CN,they should desorb spontaneously and finish the reaction cycle. The rate–limiting step for the TER mechanism is the dissociative adsorption of O2,and the reaction energy of Co@CN is 0.42 eV.The result indicates that the CO oxidation on Co@CN through the TER mechanism is superior to Pd@CN (0.48 eV). For 2Co@CN,as shown in Fig. 6, the adsorbed O2molecule reacts with two CO molecules simultaneously and produces an OCO–Co–OCO intermediate.[45]The reaction barrier is 0.38 eV, a bit smaller than that of Co@CN.
4. Conclusion
We have investigated the electronic structure of one and two Co atoms implanted on CN monolayer and their catalytic role played in CO oxidation by first-principles calculations.Our results show that the large binding energy and high diffusion barrier ensure that Co atoms are steadily anchored on CN and hard to form clusters,which are beneficial for the reactions. The adsorption energies of CO and O2on both catalysts are comparable, and the reactants molecules can be effectively captured and activated. Via three typical reaction mechanisms, we find that CO oxidation can favorably be in progress over both catalysts,and the TER mechanism is more preferable with a fairly small rate-limiting reaction barrier.
猜你喜欢
Chinese Physics B(2022年6期)2022-06-29 08:53:46
知音海外版(下半月)(2020年10期)2020-11-23 01:48:42
装备环境工程(2020年2期)2020-03-23 06:44:06
知音·上半月(2019年11期)2019-11-21 04:50:38
东方少年·阅读与作文(2019年10期)2019-10-30 09:03:41
派出所工作(2018年6期)2018-09-10 23:01:56
派出所工作(2017年6期)2017-05-30 10:48:04
派出所工作(2016年2期)2016-05-30 05:20:20
知音海外版(上半月)(2016年4期)2016-04-20 16:59:21
Chinese Journal of Chemical Engineering(2012年5期)2012-03-22 10:09:50

- Chinese Physics B的其它文章
- Coarse-grained simulations on interactions between spectrins and phase-separated lipid bilayers∗
- Constraints on the kinetic energy of type-Ic supernova explosion from young PSR J1906+0746 in a double neutron star candidate∗
- Computational model investigating the effect of magnetic field on neural–astrocyte microcircuit∗
- Gas sensor using gold doped copper oxide nanostructured thin films as modified cladding fiber
- Exact explicit solitary wave and periodic wave solutions and their dynamical behaviors for the Schamel–Korteweg–de Vries equation∗
- Suppression of ferroresonance using passive memristor emulator