
Interplay between nuclear factor erythroid 2 -related factor 2 and inflammatory mediators in COVID-19 -related liver injury
2021-06-24 01:30DanDanZhuXueMeiTanLiQingLuSiJiaYuRuLiJianXinFangLiangYiXuanLiaoWeiFanLuciaBarbierTorresAustinYangHePingYangTingLiu
World Journal of Gastroenterology 2021年22期
Dan-Dan Zhu, Xue-Mei Tan, Li-Qing Lu, Si-Jia Yu, Ru-Li Jian, Xin-Fang Liang, Yi-Xuan Liao, Wei Fan, Lucia Barbier-Torres, Austin Yang, He-Ping Yang, Ting Liu
Abstract Coronavirus disease 2019 (COVID-19 ) caused by severe acute respiratory syndrome coronavirus 2 is a global pandemic and poses a major threat to human health worldwide. In addition to respiratory symptoms, COVID-19 is usually accompanied by systemic inflammation and liver damage in moderate and severe cases. Nuclear factor erythroid 2 -related factor 2 (NRF2 ) is a transcription factor that regulates the expression of antioxidant proteins, participating in COVID-19 -mediated inflammation and liver injury. Here, we show the novel reciprocal regulation between NRF2 and inflammatory mediators associated with COVID-19 -related liver injury. Additionally, we describe some mechanisms and treatment strategies.
Key Words: COVID-19 -related liver injury; Nuclear factor erythroid 2 -related factor 2 ;Inflammatory mediator; Oxidative stress; Therapeutic targets
INTRODUCTION
Coronavirus disease 2019 (COVID-19 ), a disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2 ), was declared to be a global pandemic by the World Health Organization in 2020 . COVID-19 is caused by infection with SARS-CoV-2 , which has a single-stranded RNA approximately 26 -32 kb in length and belongs to the same coronavirus family as Middle East respiratory syndrome coronavirus and SARS-CoV-1 [1 ,2 ].
Viral toxicity, body immunity, and the induced inflammatory response all affect the occurrence and progression of COVID-19 [3 ]. The commonly accepted method of transmission is that SARS-CoV-2 binds to the receptor angiotensin-converting enzyme 2 (ACE2 ) on cells through its spike (S) glycoprotein protein, which has two domains,S1 and S2 , with the former binding the peptidase domain of ACE2 , called the receptorbinding domain, and the latter catalyzing membrane fusion to release genetic material into the cell[4 ,5 ]. After entry, SARS-CoV-2 interferes with the host’s immune defenses,evades host immune surveillance, and rapidly replicates. The replication of the virus activates monocytes, macrophages and granulocytes, accompanied by the release of many reactive oxygen species (ROS) and inflammatory cytokine storms, which eventually lead to tissue inflammatory cell infiltration, necrosis, and fibrosis[3 ,6 ].
The pathological mechanism of SARS-CoV-2 infection is very complex and unclear.It is now widely believed to be associated with the host’s immune response, the inflammatory response, and oxidative stress[3 ]. It manifests with moderate to severe respiratory symptoms and is usually accompanied by multiple organ dysfunction syndrome (MODS)[7 ]. Based on the pathogenesis of COVID-19 , it has been hypothesized that SARS-CoV-2 might directly bind to the receptor ACE2 , which is widely distributed among body tissues, such as liver tissue, and is considered a target for SARS-CoV-2 entry, which causes inflammation, oxidative stress, and proapoptotic reactions, ultimately leading to liver injury[5 ]. SARS-CoV-2 can directly cause bile duct epithelial cell dysfunction and affect hepatocyte regeneration and the immune response[8 ]. Liver dysfunction is a common complication and mainly manifests as elevated transaminase and bilirubin levels[9 -12 ]. According to statistics, 5 .1 %-69 .8 % of patients with COVID-19 suffer from liver dysfunction, occurring more commonly in patients with other severe symptoms[13 ,14 ], and 2 .1 %-4 .1 % of patients with COVID-19 undergo pre-existing liver disease[14 ]. Interestingly, liver dysfunction appears earlier in COVID-19 -related MODS, most likely because of the number of Kupffer cells,natural killer (NK) cells and NK T cells. The expression of endothelial adhesion molecules is higher in the liver than in other organs[15 ], which demonstrates that the inflammatory response, oxidative stress, and immune response may play important roles in COVID-19 -related liver injury. Moreover, SARS-CoV-2 -induced cytotoxicity,ischemia, drug toxicity, and triggering of pre-existing liver diseases are also possible contributors to the pathogenesis of COVID-19 -related liver injury[16 ,17 ]. However, the exact mechanism of COVID-19 -related liver injury remains unclear.
Nuclear factor erythroid 2 -related factor 2 (NRF2 ) is a nuclear transcription factor that is activated by oxidative stress and belongs to the family of Cap'n'Collar family of basic leucine zippers[18 ,19 ]. Kelch-like ECH-associated protein-1 (KEAP1 ), a redoxregulated substrate adaptor protein, which interacts with the Cul3 -dependent ubiquitin ligase complex[20 ]. The antioxidant response element (ARE), an electrophilic response element, is a cis-regulatory element[21 ]. NRF2 is recognized as a regulator of oxidative stress, and KEAP1 -NRF2 -ARE is one of the most important pathways of antioxidant and anti-inflammatory element signaling[22 ]. Under homeostasis, NRF2 binds to KEAP1 and is anchored to the cytoplasm[23 ]. When cells are attacked by ROS or other stimuli, NRF2 dissociates from KEAP1 and is quickly translocated to the nucleus, forming a heterodimer with the transcriptional regulator and binding to an ARE[24 ]. The complex ultimately activates the expression of antioxidant enzymes and thus leads to the removal of excessive ROS in the body, thereby exerting antioxidant effects[25 ]. Then, NRF2 is degraded in the nucleus through the β-transducin repeatcontaining protein-glycogen synthase kinase 3 (β-TrCP-GSK3 ) axis or by translocating to the cytoplasm and becoming rapidly degraded by KEAP1 [26 ]. Since the outbreak of COVID-19 , continuous studies have shown that enhancing immunity, establishing resistance to the inflammatory response, and reducing oxidative stress are keys to COVID-19 treatment, while NRF2 may be an important target for COVID-19 treatment and plays a protective role because of its dual anti-viral and anti-inflammatory properties [27 ]. Currently, NRF2 activators, such as 4 -octyl-itaconate (4 -OI) and dimethyl fumarate (DMF), can be used for the treatment of COVID-19 [28 ]. As most liver injury is associated with increased oxidative stress and an overloaded antioxidant defense system, NRF2 may also be an ideal therapeutic target for treating chronic liver disease. Indeed, NRF2 has previously been shown to protect liver cells in viral hepatitis[29 ,30 ], drug-induced liver injury[31 -33 ], cholestasis liver injury[34 -36 ], fatty liver[37 ,38 ], and other liver diseases by reversing insulin resistance, inhibiting gluconeogenesis and lipid accumulation, and enhancing the anti-inflammatory and antioxidative effects[39 ]. We speculate that NRF2 may also play a regulatory role in COVID-19 -related liver injury. In this review, we systematically describe the reciprocal regulation between NRF2 and inflammatory mediators in COVID-19 -related liver injury.
NRF2 AND INFLAMMATORY MEDIATORS
Cytokines are small-molecule messengers secreted by cells, and they can exert effects on the same type of cell in the organ periphery and cells in other organs[40 ]. Most cytokines are in a lytic state, and some exist in membrane form. Cytokines can be secreted by most cells and possess a wide range of functions. Interleukins (ILs) are a group of cytokines that were first seen to be expressed by white blood cells.Chemically attractive cytokines are called chemokines. Cytokines that interfere with viral replication are called interferons (IFNs).
When the body is stimulated by endogenous or exogenous stimuli such as viruses, it usually triggers the immune defense system and oxidative stress response to maintain homeostasis. This process is often accompanied by the production of many cytokines[41 ], which interfere with mitochondrial function and lead to the production of a large amount of ROS, thereby inducing the expression of IL-1 , IL-6 , tumor necrosis factor alpha (TNF-α), IL-8 receptor-β (CXCR2 ), monocyte chemotactic protein-1 , and cell adhesion molecule 1 and promoting the aggregation of macrophages and chemotactic neutrophils[42 -44 ]. Then neutrophils produce myeloperoxidase and catalyze the production of large amounts of hypochlorous acid, which kills cells through strong oxidation reactions[45 ]. Cell injury induces the expression of high mobility group box 1 , osteopontin, and Toll-like receptor 4 , recruiting more neutrophils, amplifying the inflammatory response, and further aggravating tissue injury[46 ].
It is thought that COVID-19 patients suffer from cytokine storm symptoms and an uncontrolled release of proinflammatory cytokines. The levels of proinflammatory factors such as IL-1 β, IL-7 , IL-8 , IL-9 , IL-10 , TNF-α, and other inflammatory factors are higher in COVID-19 patients than in healthy adults[47 ,48 ]. Serum inflammatory cytokine levels are positively correlated with indicators of liver dysfunction in patients, suggesting potential mechanisms between liver injury and the inflammatory response[49 ]. NRF2 negatively regulates the expression of these inflammatory cytokines. For example, in human macrophages, NRF2 can inhibit the expression of inflammatory cytokines, such as IL-1 β, IL-6 , and TNF-α, by blocking the recruitment of RNA polymerase II, thus reducing the inflammatory response. Moreover, an agonist of NRF2 represses the replication of SARS-CoV-2 and the inflammatory response[50 ].
The NRF2 -activating compound resveratrol is a growth suppressor and apoptosis promoter in liver cancer cells[51 ]. Resveratrol can reduce the expression of IL-1 β.Another NRF2 -activating compound, PB125 , which contains the active ingredients carnosol, withaferin A, and luteolin, significantly downregulates IL-1 β, IL6 , TNF-α,intercellular adhesion molecule 1 , vascular cell adhesion molecule 1 (VCAM1 ), Eselectin, and IFN-γ-induced gene expression[52 ]. The inflammatory response and oxidative stress are the main pathological features of COVID-19 , and disease progression has been related to redox imbalance. The NRF2 antioxidant pathway is suppressed in COVID-19 patients and cells infected with SARS-CoV-2 show activated GSK-3 , which is a conserved serine/threonine kinase that degrades NRF2 [53 ]. The NRF2 agonists 4 -OI and DMF can inhibit the replication of SARS-CoV-2 [28 ]. All of these findings support the idea that NRF2 has dual antiviral and anti-inflammatory properties and may be a potential therapeutic target for the treatment of COVID-19 .
Pre-existing liver disease may play an important role in COVID-19 -related liver injury, since an investigation found that a small proportion of patients with COVID-19 also have pre-existing liver diseases[54 -56 ]. This effect may be due to the significant decrease in the number of lymphocytes, CD4 + T cells, CD8 + T cells, B cells, and NK cells in patients with COVID-19 , which prevents a proper immune response[57 ],ultimately leading to the recurrence of pre-existing liver disease. Glucocorticoids effectively reduce mortality in patients with severe COVID-19 [58 ]. In addition to its anti-inflammatory function, glucocorticoids also have immunosuppressive effects,which can also participate in the reactivation of pre-existing liver diseases.
As shown in Figure 1 , inflammatory molecules are involved in the interactions with NRF2 in viral infection, as listed in a dataset generated by ingenuity pathway analysis(IPA). They include IL1 A, IL1 B, IL4 , IL5 , IL6 , IL10 , IL13 , IL19 , IL36 G, IL1 RN, IFNGR2 ,IFNB1 , CCL2 , CYSLTR1 , EXCR3 , CXCL8 , CXCL6 , CXCL3 , CXCL2 , CXCL10 , CCL5 ,CCL4 , CCL20 CCL11 , TGF-β, TNFRSFA, TNF, and MST1 R. Current literature shows that these molecules are related to the COVID-19 -mediated inflammatory response[41 -48 ].
INTERPLAY OF NRF2 /NF-κB AND INFLAMMATORY MEDIATORS IN COVID-19 -RELATED LIVER INJURY
Activated nuclear factor kappa-B (NF-κB) is a key transcription factor in the inflammatory response and oxidative stress. The NF-κB signal transduction pathway is a typical proinflammatory pathway[59 ]. There are five members in the NF-κB family:p50 (NF-κB1 ), p52 (NF-κB2 ), p65 (RelA), RelB, and c-Rel[60 ]. Under physiological conditions, NF-κB and inhibitor kappa B (IκB) combine and are maintained in the cytoplasm in an inactive state. Upon viral infection or other stimuli, IκB is phosphorylated under the action of IκB kinase (IKK) and is then ubiquitinated by the β-TrCPSkp1 -Cullin1 pathway, resulting in the release and nuclear translocation of NF-κB,inducing the production of a large number of inflammatory mediators, and leading to oxidative stress and inflammatory cascades, thereby affecting cell survival, mutation,and proliferation[61 ]. According to current studies, NRF2 plays a beneficial role mainly by regulating redox metabolism through interactions with NF-κB and regulating proinflammatory genes[62 -64 ]. Several studies have shown reciprocal regulation between NRF2 and NF-κB in inflammatory diseases[65 ]. Moreover, the activation of NF-κB can promote the release of different types of cytokines, such as IL-1 , IL-2 , IL-6 , IL-12 , TNF-α and granulocyte-macrophage colony stimulating factor,which can directly participate in acute and chronic inflammation of the liver (Figure 2 ),causing liver injury[60 ]. Among these cytokines, TNF-α is a cytokine mainly produced by macrophages/monocytes and is involved in the gene expression of growth factors,cytokines, transcription factors, and receptors[66 ]. There are two types of TNF-α receptors: TNF receptor 1 (TNFR1 ) and TNF receptor 2 (TNFR2 ). TNF-α can activate NF-κB through the TNFR1 -NF-κB signaling axis[66 ], and in turn, NF-kB promotes the expression of inflammatory cytokines[60 ], thereby forming a negative cycle that further feeds the destructive cytokine storm and aggravates damage to the liver tissue[36 ,67 ,68 ]. According to previous studies, lipopolysaccharides can promote NF-κBand NRF2 -mediated responses[69 ]. The initial proinflammatory response is driven by NF-κB[60 ], and when the activity of NRF2 reaches its maximum level, NF-κB activation is inhibited. NF-κB subunits p50 and p65 can induce NRF2 transcription by binding specific sites of NRF2 , and NRF2 can inhibit the expression of NF-kB in turn upon IκB dephosphorylation[62 ]. CBP-p300 , a transcriptional auxiliary activator, can acetylate nonhistone proteins, such as NRF2 and p65 , the lysine residues of which are equipped with acetyl groups, enhancing gene transcription[70 -72 ]. NRF2 has 605 amino acid residues and six conserved domains, namely, Neh1 -Neh6 [18 ,19 ]. CBP can bind to Neh4 and Neh5 of NRF2 , resulting in acetylation of the Neh1 domain. In addition, CBP can bind to phosphorylated p65 at Ser276 . Overexpressed p65 can competitively bind to CBP, thereby inhibiting the transcriptional activity of NRF2 but enhancing the transcriptional activity of NF-κB promoter genes[70 ].

Figure 1 An interaction network analysis (ingenuity pathway analysis) was performed to predict nuclear factor, erythroid 2 -like 2 -mediated inflammatory cytokine-related genes in viral infections. Solid lines indicate direct regulation, and dotted lines depict indirect interactions.ABCC2 : ATP-binding cassette subfamily C member 2 ; ACE: Angiotensin I-converting enzyme; ACE2 : Angiotensin I-converting enzyme 2 ; C5 : Complement C5 ; Ccl2 :Chemokine (C-C motif) ligand 2 ; CCL: C-C motif chemokine ligand; CXCR3 : C-X-C motif chemokine receptor 3 ; CYSLTR1 : Cysteinyl leukotriene receptor 1 ; F10 :Coagulation factor X; IFNB1 : Interferon beta 1 ; IFNG: Interferon gamma; IFNGR2 : Interferon gamma receptor 2 ; IL: Interleukin; IL1 A: Interleukin 1 alpha; IL1 B:Interleukin 1 beta; IL1 RN: Interleukin 1 receptor antagonist; IL36 G: Interleukin 36 gamma; MST1 R: Macrophage-stimulating 1 receptor; NFE2 L2 : Nuclear factor,erythroid 2 -like 2 ; PF4 : Platelet factor 4 ; PIP5 K1 C: Phosphatidylinositol-4 -phosphate 5 -kinase type 1 gamma; SPP1 : Secreted phosphoprotein 1 ; TGF-beta:Transforming growth factor beta; TNF: Tumor necrosis factor; TNFRSF1 A: TNF receptor superfamily member 1 A; VCAM1 : Vascular cell adhesion molecule 1 .
KEAP1 is the intermediate link between NRF2 and NF-κB. ARE is a cis-acting element, and the NRF2 -ARE pathway is the most important endogenous antioxidant excitation pathway[73 ]. On one hand, p65 can increase the nuclear level of KEAP1 to inhibit the NRF2 -ARE pathway, thus reducing the expression of cytoprotective enzymes. On the other hand, KEAP1 can initiate the autophagic degradation of inhibitor of NF-κB kinase subunit β (IKKβ) by preventing the binding of heat shock protein 90 (HSP90 ) to IKKβ. Additionally, KEAP1 can specifically bind to IKKβ and then connect with the ubiquitin connexin CUL3 -RBX1 complex, which promotes IKKβ degradation and inhibits the expression of NF-kB[74 ].
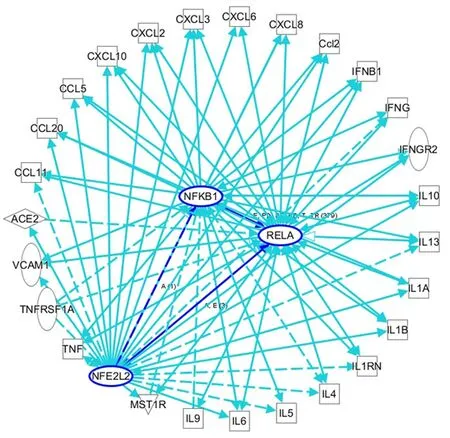
Figure 2 An ingenuity pathway analysis was performed to predict nuclear factor, erythroid 2 -like 2 /nuclear factor kappa B-mediated inflammatory cytokine-related genes in viral infections. Solid lines indicate direct regulation, and dotted lines depict indirect interactions. ACE: Angiotensin I-converting enzyme; Ccl2 : Chemokine (C-C motif) ligand 2 ; CXCL: C-X-C motif ligand; IL: Interleukin; IL1 B: Interleukin 1 beta; IFNG: Interferon gamma; IL1 RN:Interleukin 1 receptor antagonist; NFKB1 : Nuclear factor kappa B subunit 1 (p50 ); RELA: RELA proto-oncogene, NF-κB subunit (p65 ); TNF: Tumor necrosis factor;TNFRSF1 A: TNF receptor superfamily member 1 A; VCAM1 : Vascular cell adhesion molecule 1 .
COVID-19 is always accompanied by an increase in inflammatory cells and proinflammatory factors. Oxidative stress is an important mechanism for the occurrence and development of COVID-19 , and is closely related to the prognosis of the disease[12 ]. ROS are the key agents in oxidative stress; some researchers propose monitoring ROS levels as diagnostic criteria for COVID-19 [75 ]. As a key regulator of oxidative stress and inflammation, NF-κB plays an important role in the progression of COVID-19 . In patients with COVID-19 , a severe systemic inflammatory response is always followed by acute respiratory distress syndrome (ARDS) and sepsis, which leads to tissue ischemia and hypoxia and the generation of ROS, thus activating the NF-κB pathway[76 ]. In addition, Hirano et al[77 ] proposed that ACE2 is the key factor in the early stage of COVID-19 , and the IL-6 -signal transducer and activator of transcription 3 (STAT3 ) axis plays an important role in the inflammatory cytokine storm in the late stage of the disease. ACE2 is necessary for SARS-CoV-2 entry, and it can also inactivate angiotensin 2 (AngII), which can play a proinflammatory roleviathe angiotensin 2 receptor (AT1 R). SARS-CoV, has high homology with SARS-CoV-2 and binds to ACE2 on the cell membrane, which results in an increase in AngII in serum[78 ]. In animal models, AT1 R inhibitors can prevent ARDS caused by SARS-CoV infection[78 ]. The AngII-AT1 R axis also activates NF-κB and ADAM metallopeptidase domain 17 (ADAM17 ), which can promote the production of an epidermal growth factor ligand (EGFR) ligand and TNF-α, and this EGFR ligand and TNF-α can also stimulate NF-κB[79 ]. ADAM17 can also activate STAT3 via IL-6 , thus activating NF-κB[80 ]. The NF-κB-triggered positive feedback loop for IL-6 signaling in type 1 collagen+nonimmune cells is termed an IL-6 amplifier. Indeed, it has been proven that both the NF-κB pathway and STAT3 pathway are activated in COVID-19 , promoting an increase in proinflammatory factors and inflammatory cells by activating the IL-6 amplifier, thus forming a vicious cycle and eventually leading to a systemic inflammatory response and MODS[80 ]. Among the various organs affected by MODS,COVID-19 -related liver injury has a high incidence, ranging from 5 .1 % to 69 .8 %according to recent reports, and manifests mainly as elevated transaminase and bilirubin levels (Table 1 ). Liver injury greatly affects the progression and prognosis of COVID-19 . However, the exact mechanisms of liver injury are unclear due to limited data. In addition to the application of drugs that cause liver injury, direct viral cytotoxicity and reactivity of existing liver disease, systemic inflammatory response,ischemia, and hypoxia may play crucial roles, as increased oxidative stress and an overloaded antioxidant defense system have always been acknowledged as key factors in the occurrence and development of various liver injuries[81 ]. Oxidative stress can stimulate Kupffer cells to produce cytokines that promote the apoptosis of hepatocytes and can also induce stellate cell activation/proliferation and collagen synthesis to promote fibrosis and cirrhosis[82 ]. To maintain redox homeostasis, the liver fights oxidative stress through a complex antioxidant system. NRF2 is associated with enhancement of the endogenous antioxidant system and has been identified as a target to prevent liver injury, inhibit liver fibrosis, and promote liver regeneration[74 ,83 -86 ].As shown through IPA analyses of the interactions among NRF2 , NF-κB, and inflammatory cytokines in viral infections, the interaction and balance between NRF2 and NF-kB may play important roles in COVID-19 -related liver injury, and targeting their interactions may be an important therapeutic strategy for COVID-19 treatment.
NRF-HO1 INTERPLAY IS AN IMPORTANT TARGET FOR INFLAMMATORY MEDIATORS IN COVID-19 -RELATED LIVER INJURY
Glutathione (GSH) is an important molecule involved in redox homeostasis, and NRF2 promotes its synthesis by regulating glutamate-cysteine ligase catalytic subunit(GCLC), glutamate-cysteine ligase regulatory subunit (GCLM), glutamine synthetase(GS or GSr1 ), GSH reductase (GR), GSH peroxidase (GPx), and GSH S-transferases(GSTs)[92 ]. The tricarboxylic acid cycle (TCA cycle) is the central link and cross hub of glucose metabolism, lipid metabolism, and protein metabolism. NRF2 participates in the TCA cycle by regulating the expression of glucose 6 -phosphate dehydrogenase(G6 PD), malic enzyme 1 , and isocitrate dehydrogenase (IDH), which are involved in the production of NADPH, a key cofactor in promoting antioxidant reactions[93 -95 ].NRF2 can also affect intermediate metabolism, increases the availability of substrates for the mitochondrial respiratory chain, and increases nicotinamide adenine dinucleotide phosphate (NAPDH) production through crosstalk between the glycolysis and pentose phosphate pathways[96 ]. In addition, under oxidative stress or starvation, NRF2 helps maintain mitochondrial homeostasis, clears oxidized or damaged proteins and organelles and removes excessive ROS, providing a favorable environment for cell survival[97 ]. In conclusion, NRF2 can play roles in antioxidative stress and cell protection by maintaining redox homeostasis in the body.
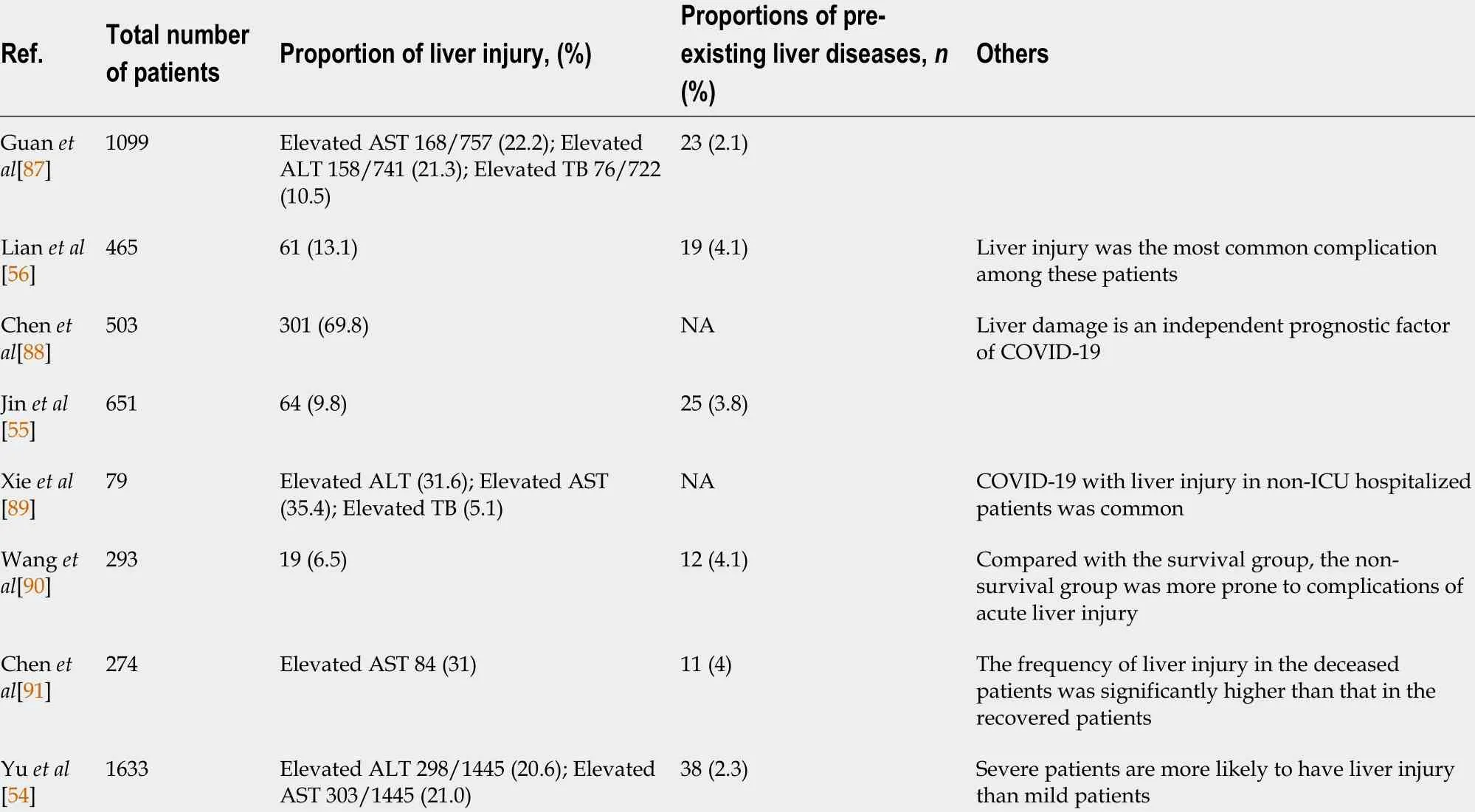
Table 1 Characteristics of liver injury during coronavirus disease
Many researchers have proposed the hypothesis that COVID-19 -related liver damage may be caused by systemic inflammatory response to viral infection. After SARS-CoV-2 infection and the increased production of ROS[98 ], the inflammation response of the body is activated and the expression of proinflammatory cytokines including IL-1 β, IL-6 , IL-8 , and TNF-α is greatly increased, and immune cells are continuously activated and are accumulating[99 ,100 ]. The combined effects of many immune cells and proinflammatory factors lead to a cytokine storm[4 ,99 -102 ], which may further result in multiple organ function damage, including liver dysfunction,and even the death of the patient[103 ,104 ]. Moreover, there seems to be a positive correlation between the severity of COVID-19 and the level of inflammation [57 ,100 ,105 -107 ], and the incidence of liver damage is related to the severity of COVID-19 [13 ],which also reveals the close relationship between COVID-19 -related liver injury and the inflammatory response. Since many cases of limb[108 ], gastrointestinal[109 ], heart[110 ], and cerebral[111 ] ischemia have been reported, it is reasonable to suspect that tissue ischemia is among the causes of COVID-19 -related liver injury, in which the inflammatory response may cause local blood stasis, vascular endothelial destruction and excessive activation of the coagulation system, leading to local thrombosis and tissue ischemia, ultimately resulting in liver injury. Moreover, severe inflammatory reactions, such as sepsis[76 ], which cause hemodynamic instability and lead to ischemia, may also be among the causes of liver injury. Strong evidence points to the major causes for liver injury involve the systemic inflammatory response and oxidative stress following SARS-CoV-2 infection.
Regarding the mechanisms of direct damage and inflammation-induced damage after SARS-CoV-2 infection, heme oxygenase 1 (HO-1 ) seems to be involved and could represent a therapeutic strategy. HO-1 can inhibit the replication of influenza A virus,human respiratory syncytial virus, and hepatitis B virus, among others. In all cases,viral replication blockade is achieved through the type I IFN response mediated by HO-1 [112 ]. Therefore, it is reasonable to assume that HO-1 inhibits SARS-CoV-2 replication by the same mechanism. HO-1 and its metabolites have significant antiinflammatory effectsviaNRF2 . HO-1 metabolizes free heme into carbon monoxide(CO), biliverdin and iron, exhibiting cell protection and anti-apoptosis and immune regulation properties[113 ]. In animal models of sepsis and renal ischemia-reperfusion injury with significant systemic inflammatory responses, HO-1 showed great antiinflammatory effects and its overexpression provided cell protection[112 ]. Hence, it can be speculated that HO-1 counteracts the cytokine storm produced by COVID-19 .Additionally, the CO product of HO-1 can stimulate the release of hydrogen sulfide(H2S)[114 ]. H2 S is a natural defense against enveloped RNA virus infections that can activate regulatory T cells, maintain the body’s immune homeostasis, and exert antiviral effects[114 ]. H2 S can also oversulfate the KATP channel on the white blood cell membrane to keep it open, preventing white blood cells adherence to the endothelium and triggering the inflammatory cascade[114 ]. On the one hand, H2 S regulates endothelial cell function and attenuates ischemia. On the other hand, H2S controls the body’s inflammatory response and prevents liver injury. Therefore, HO-1 activated by NRF2 can inhibit viral replication to control viral infection and can also balance host defenses through the HO-1 /CO/H2 S axis, protecting endothelial structures, improving ischemia, and protecting the liver from damage induced by SARS-CoV-2 infection.
Figure 3 shows reciprocal regulation between NRF2 /redox-related genes and inflammatory mediators. In particular, upregulation of NRF2 /HO-1 (HMOX1 ) may be an important strategy for the treatment of the liver damage caused by COVID-19 .
NRF2 -MYC IS A KEY TARGET FOR INFLAMMATORY MEDIATORS IN COVID-19 -RELATED LIVER INJURY
NRF2 consists of seven highly conserved domains, NRF2 -ECH homology 1 (Neh1 ) to Neh7 . Each of the Neh domains executes distinct functions[18 ,19 ]. Neh1 is a CNC-bZIP domain that allows NRF2 to heterodimerize with small musculoaponeurotic fibrosarcoma (MAF) proteins (MAFF, MAFG, and MAFK)[115 ]. Under normal physiological conditions, NRF2 is degradedviathe ubiquitin-proteasome pathway in a KEAP1 -dependent manner and maintained at a basal level[36 ]. Under stress conditions, the cysteine residues in KEAP1 are modified when they are oxidized,causing the loss of its adaptor activity[20 ]. Then NRF2 is disengaged from the regulatory complex, is phosphorylated, and accumulates in the nucleus, where it heterodimerizes with small MAF proteins and regulates ARE-driven genes[115 ]. These proteins regulate transcription when they dimerize among themselves[116 ,117 ]; that is,they seem to serve as transcriptional activators by dimerizing with other basic zipper proteins such as NFE2 L2 [115 ]. The interaction of MAFG and NFE2 L2 results in activation of the ARE, which is present at the promoter-proximal region of many genes involved in the antioxidant defense and triggers the expression of certain genes[115 ].MAFG expression is induced by oxidative stressors, such as hydrogen peroxide and electrophilic compounds, increasing NRF2 and GSH expression and activating the NRF2 -ARE pathway[118 ]. NRF2 exerts protective effects on various liver diseases [119 ,120 ]. Therefore, NRF2 can also be a therapeutic target and NRF2 activators can also be used for the treatment of COVID-19 -related liver injury. MAFG, as a molecular chaperone is required for the function of NRF2 , may also be a target for the treatment of COVID-19 -related liver injury.
C-MYC is a family of proto-oncogenes and encodes a nuclear phosphoprotein that plays roles in cell cycle progression, apoptosis, and cell transformation[121 ]. MNT is a MAX network transcriptional repressor[122 ]. C-MYC and MNT each have a basic helix-loop-helix-zipper domain (bHLHzip) with which they bind the canonical DNA sequence CACGTG, known as an E-box, following heterodimerization with MAX proteins. The protein complexes bind to the E-box DNA sequence and regulate the transcription of specific target genes[122 ]. The MYC-MAX and MNT-MAX networks comprise transcription factors that regulate gene-specific transcription[122 ]. The heterodimer MNT-MAX is a transcriptional repressor, and the heterodimer MYCMAX is a transcriptional activator[123 -125 ]. We also reported that c-MYC can interact with NRF2 to repress NRF2 -mediated transactivation of the ARE[126 ]. Their interaction may regulate the oxidative stress pathway. An IPA showed the interactions among NFE2 L2 /MAF/MYC/MNT and inflammatory mediators. NRF2 /MYC/MNT/MAFG are involved with inflammatory mediators IL10 , IL1 A, IL1 B, IL36 G, IL4 , IL5 ,IL6 , IFNG, IFNG, CXCL8 , VCAM1 , TNF, TGF-β, SPP1 and MST1 R (Figure 4 ).

Figure 3 An ingenuity pathway analysis was performed to predict NRF2 /redox-related genes and inflammatory cytokine-related gene expression in viral infections. Solid lines indicate direct regulation, and dotted lines depict indirect interactions. Ccl2 : Chemokine (C-C motif) ligand 2 ; CXCL: CX-C motif ligand; GCLC: Glutamate-cysteine ligase catalytic subunit; GCLM: Glutamate-cysteine ligase modifier subunit; GPX1 : Glutathione peroxidase 1 ; HMOX1 :Heme oxygenase 1 (HO-1 ); IFN: Interferon; IL: Interleukin; TNF: Tumor necrosis factor; TNFRSF1 A: TNF receptor superfamily member 1 A; VCAM1 : Vascular cell adhesion molecule 1 .
Upon analyses of a protein-protein interaction network with 65 intersecting targets,c-MYC was found to be one of the core targets for COVID-19 treatment[127 ]. MAFG and c-MYC are upregulated and MNT is downregulated in liver injury[125 ,128 ]. MAX directly interacts with the c-MYC, MAFG, MNT, and NRF2 proteins, whereas c-MYC and MAFG interact with NRF2 . As shown in Figure 5 , these complexes bind to the Ebox region in the promoters of the c-MYC and MAFG genes, leading to gene activation. The complexes can also bind to AREs, leading to the dysregulation of the following genes: TXN-related genes (TXN, TXNrd1 , and Srxn1 ); ROS and xenobiotic detoxification-related genes (GSTA1 /2 and GSTM1 /2 /3 ), GSTP1 , and Nqo1 ); heme and ion metabolism-related genes (HMOX1 or HO-1 , Ft1 , and Fth); NADPH regeneration-related genes (G6 pD, PgD, IDH1 and Me1 ); and GSH production-related genes (XCT, GCLC, GCLM, and GSR1 ). This gene dysregulation may lead to COVID-19 -related cytokine storm and liver injury.
SEVERITY OF LIVER INJURY IN TERMS OF ADVERSE OUTCOMES OF SARS-COV-2 INFECTION
Alanine aminotransferase (ALT) was selected to represent liver injury rather than aspartate aminotransferase (AST) because the sources of extrahepatic AST are very common, including the heart and skeletal muscle during related muscle breakdown.Therefore, in patients infected with SAR-CoV-2 , we classified liver damage into normal/mild (< 2 fold the upper limit of normal, upper limit of normal [ULN]),moderate (2 - to 5 -fold ULN), and severe (> 5 fold ULN) according to the degree of ALT elevation (Table 2 )[129 ].
Normal and mild liver injury account for the largest proportion (60 %-90 %) of SARCoV-2 -infected patients. Severe liver injury accounts for less than 20 % of these patients. In addition, moderate to severe liver injury is more common in patients requiring intensive care unit (ICU) care. However, in general, the most common liver injury is mild liver injury, and the degree of liver injury is independently related to the occurrence of end events (ICU admission, death, mechanical injury)[129 -131 ]. Only 5 %of COVID-19 patients acquire severe liver damage[132 ]. The Cai et al[133 ] studyshowed that the incidence of liver injury in hospitals was higher than that at the time of admission. For these patients, few other factors affected liver test abnormalities,such as potential liver disease or drug use. Therefore, it can be speculated that the occurrence of liver damage is caused by COVID-19 . Zhao et al[134 ] compared patients with mild COVID-19 systems with non-infected patients. The absolute value of lymphocyte level differences in the two groups was low, and there was no difference in C-reactive protein or IL-6 level. ALT was not elevated in patients with pneumonia unrelated to COVID-19 when they were admitted to the hospital, but it was elevated in 28 % of COVID-19 patients. Compared with the general inflammation caused by other pathogens, the specific inflammation caused by COVID-19 is more likely to cause abnormal liver function. Additionally, severe liver damage caused by SARS-CoV-2 infection is associated with serum inflammatory markers (white blood cell count and neutrophil-to-lymphocyte ratio are significantly higher). Age, hypertension, and the presence of diabetes are negatively correlated with severe liver damage in COVID-19 patients[129 ]. It can be speculated that young patients may have a stronger immune and inflammatory response to infection, which may lead to more severe liver damage.In other words, the inflammatory response can cause liver damage and determine the degree of liver damage. Therefore, inflammation plays an important role in the development of liver injury induced by COVID-19 . Because of the individual response to the virus, different patients may exhibit different degrees of liver injury; therefore, it is necessary to grade the degree of liver injury. We described the anti-inflammatory effect of NRF2 above, which is of great benefit to the prevention or treatment of COVID-19 -mediated liver injury, and is also crucial for improving the prognosis of patients.

Table 2 Classification of the severity of liver injury in terms of the adverse outcome of coronavirus disease

Figure 4 An ingenuity pathway analysis was performed to predict that nuclear factor, erythroid 2 -like 2 /MYC proto-oncogene, bHLH transcription factor/MAX network transcriptional repressor/MAF bZlP transcription factor G regulates the corresponding inflammatory cytokines. Solid lines indicate direct regulation, and dotted lines depict indirect interactions. CXCL: C-X-C motif ligand; MAFG: MAF bZIP transcription factor G;MAX: MYC-associated factor X; MNT: MAX network transcriptional repressor; MYC: MYC proto-oncogene, bHLH transcription factor; IFN: Interferon; IL: Interleukin;TGF-beta: Transforming growth factor beta; TNF: Tumor necrosis factor; TNFRSF1 A: TNF receptor superfamily member 1 A; VCAM1 : Vascular cell adhesion molecule 1 .

Figure 5 Interplay between c-MYC, MAX network transcriptional repressor, MAF bZIP transcription factor G, and nuclear factor, erythroid 2 -like 2 proteins in viral infections. MAX network transcriptional repressor (MAX) directly interacts with c-MYC and MAX network transcriptional repressor proteins, while c-MYC and MAF bZIP transcription factor G (MAFG) interact with nuclear factor, erythroid 2 -like 2 (NRF2 ). These complexes bind to E-box elements in the promoters of c-MYC and MAFG genes, regulating their activation. In addition, c-MYC and MAFG interact with NRF2 , and their complexes bind to antioxidant response elements, regulating downstream gene expression. COVID-19 : Coronavirus disease 2019 .
CONCLUSION
There are many reasons for COVID-19 -related liver injury including the inflammatory response and oxidative stress, ischemia, drug toxicity, SARS-CoV-2 cytotoxicity, and pre-existing liver disease. Regardless of the etiology, inflammatory cell infiltration, cell necrosis, and liver tissue fibrosis may be the final pathological result. As an important transcription factor of antioxidant stress and anti-inflammation, NRF2 can exert protective effects on COVID-19 -related liver injury. The KEAP1 -NRF2 -ARE, NRF2 -HO-1 , NRF2 -MYC/MNT/MAFG, and NRF2 -NF-κB pathways are potential preventive and therapeutic targets in COVID-19 -related liver injury.
ACKNOWLEDGEMENTS
We thank the authors of the primary studies for their responses to our information requests.
 World Journal of Gastroenterology2021年22期
World Journal of Gastroenterology2021年22期
- World Journal of Gastroenterology的其它文章
- Role of imaging in evaluating the response after neoadjuvant treatment for pancreatic ductal adenocarcinoma
- High fecal calprotectin levels are associated with SARS-CoV-2 intestinal shedding in COVID-19 patients: A proof-of-concept study
- Liver injury in COVID-19 : Detection, pathogenesis, and treatment
- Enhancer of zeste homolog 2 contributes to apoptosis by inactivating janus kinase 2 / signal transducer and activator of transcription signaling in inflammatory bowel disease
- Helicobacter pylori promotes invasion and metastasis of gastric cancer by enhancing heparanase expression
- Role of bile acids in liver diseases mediated by the gut microbiome