
PRKG1在前列腺癌中的表达及其机制
2021-06-20 04:46饶大庞项之洋虞海峰
温州医科大学学报 2021年6期
饶大庞,项之洋,虞海峰
温州医科大学附属第二医院育英儿童医院 泌尿外科,浙江 温州 325027
前列腺癌是第二常见的男性恶性肿瘤,也是男性癌症死亡的第四大原因[1]。亚洲男性前列腺癌的发病率和病死率在过去几十年里迅速上升,大多数患者就诊时已为晚期[2]。本课题检测cGMP依赖蛋白激酶(cGMP dependent protein kinase type I,PRKG1)在前列腺癌中的表达,研究PRKG1敲除对前列腺癌细胞的影响,为探寻前列腺癌新的治疗靶标提供思路[3]。
1 材料和方法
1.1 材料 75例前列腺癌组织和12例正常前列腺组织及相关临床资料由西安艾丽娜生物科技有限公司提供,包括PRKG1表达组织芯片免疫组化结果、前列腺癌的临床病理特征(组织病理学和组织学分级)。肿瘤分期参照美国癌症联合委员会(AJCC)第8版。病理特征评价采用Gleason评分。本研究通过温州医科大学附属第二医院医学伦理委员会审查批准(伦理审批号:2021-K-01-01)。
抗PRKG1、β-微管蛋白(β-Tubulin)、基质金属蛋白酶-9(matrix metallopeptidase 9,MMP9)、埃兹蛋白(Ezrin)、钙黏附蛋白-2(cadherin-2,CDH-2)、Bax、Bcl-2及钙黏附蛋白-1(cadherin-1,CDH-1)的一抗购自英国Abcam公司,山羊抗兔二抗(pv6001)购自北京中衫金桥公司。细胞培养试剂购自美国Gibco公司。人前列腺癌细胞系22RV1购自上海烜雅生物科技有限公司。携带Cas9和gRNA的质粒载体由杭州擎科生物科技有限公司构建及验证。CCK8试剂盒购自日本同仁化学研究所。DAB显色试剂盒20×(ZLI-9018)购自北京中衫金桥公司。
1.2 方法
1.2.1 免疫组化:组织微阵列玻片的HE染色采用标准程序进行。载玻片经二甲苯脱蜡、乙醇漂洗、重新水化。抗原修复采用枸橼酸缓冲液。将阵列浸泡在3%过氧化氢缓冲液中30 min来淬灭内源性过氧化物酶。用PRKG1一抗(1:50)孵育后,4 ℃过夜(16~18 h)。37 ℃水浴复温30 min,PBS浸泡5 min 3遍。滴加二抗,常温20 min;PBS浸泡5 min 3遍。DAB显色,随后自来水冲洗。苏木素复染20 s,自来水冲洗。浸入PBS溶液3~5 s返蓝;立即蒸馏水冲洗。依次浸泡75%乙醇、85%乙醇、95%乙醇、100%乙醇各5 min,二甲苯中各10 min脱水。切片上滴加中性树胶,盖玻片覆盖,封片。
每个标本均在光学显微镜(BX40,日本奥林帕斯)下检查,以确定组织病理学特征和抗PRKG1免疫反应性。由两位资深病理医师双盲评分,结果一致者为最后判定结果。免疫组化结果判定[4]:阳性染色定位于细胞浆,采用下述方法对免疫组化染色结果进行半定量分析:首先按阳性强度评分:0分为无色,1分为淡黄色,2分为棕黄色,3分为棕色;再将阳性细胞数评分:无阳性细胞为0分,0~10%为1分,10%~50%为2分,≥50%为3分。综合阳性强度和阳性细胞数评分:两者的乘积为最后得分,得分>5分为阳性,≤5分为阴性。
1.2.2 细胞培养:人前列腺癌细胞系22RV1培养于RPMI1640含10%胎牛血清的培养液中,37 ℃,5%CO2孵育。以每孔4×105个细胞种入6孔板中,24 h后更换培养液。用0.25%胰酶消化传代,取对数生长期细胞进行实验,显微镜下细胞计数并调整浓度。
1.2.3 CRISPR/Cas9敲除:首先利用CRISPR/Cas9技术在22RV1细胞中敲除PRKG1的表达。将已建立的菌落命名为22RV1-KO,并以22RV1细胞(22RV1-CON)为对照。用于构建人类PRKG1基因gRNA 2个引物包括PRKG1-Mus-EXON1-gRNA-F1:5"-CACCGACATCGG CCCGCGGACCACC-3"、PRKG1-Mus-EXON1-gRNA-R1:5"-AAACGGTGGTCCGCGGGCCGATGTC-3"和PRKG1-Mus-EXON1-gRNA-F2:5"-CACCGTCCTTCCACGACCTGCGCC-3’、PRKG1-Mus-EXON1-gRNA-R2:5"-AAACGGCGCAGGTCGTGGAAGGAC-3"。根据生产厂家的操作规程,将携带cas9和2个gRNA的质粒转染导入22RV1细胞,获得稳定的22RV1-KO细胞系。另外将单纯cas9的对照组质粒转染导入22RV1细胞获得稳定的22RV1-CON对照组细胞系。转染3 d后,在96孔板上以0.5个细胞/孔为单位进行限制稀释法。当细胞达到70%融合度时,用100 μmol/L PMSF的细胞裂解液裂解细胞。用Western blot法检测每个克隆的PRKG1蛋白表达。随后使用PRKG1蛋白非表达克隆进行实验。
1.2.4 细胞增殖:用CCK-8试剂盒检测细胞增殖情况。将细胞以1 000个/孔的密度接种到96孔板中。每孔加入10 μL CCK-8试剂,37 ℃,5% CO2孵育2 h。在450 nm波长处测定吸光度。
1.2.5 迁移和侵袭:迁移和侵袭通过Transwell实验检测,小室底膜的上室表面涂覆并放置在37 ℃下30 min。细胞(2×105)悬浮在500 μL无血清培养基中,加入上室。下室加入含10%胎牛血清的RPMI-1640培养液。24 h后用3.7%多聚甲醛固定膜上的浸润性细胞,结晶紫染色,在400倍显微镜下计数。细胞侵袭能力也用同样的方法进行检测,但时间设为48 h。随机选择7~10个区域来手动计数侵袭和迁移细胞的数量,并计算平均值。
1.2.6 Western blot:收集细胞加入含有100 μmol/L PMSF的细胞裂解液,4 ℃、13 500 r/min离心10 min,取上清液采用BCA法测定蛋白浓度。取30 μg蛋白,经10% SDS-PAGE电泳分离后电转至PVDF膜上,5%脱脂牛奶室温封闭1 h后,分别加入抗MMP9(1:1 000)、Ezrin(1:1 000)、CDH-2(1:1 000)、Bax(1:1 000)、Bcl-2(1:1 000)、CDH-1(1:1 000)和GAPDH(1:1 000)4 ℃过夜,洗膜后,加入二抗室温温育1 h。ECL化学显色,ImageJ(v1.8.0)软件分析蛋白条带。实验重复3次。
1.3 统计学处理方法 采用SPSS26.0软件进行统计分析,所有实验结果重复3次。计量资料采用±s表示,2组间比较采用独立t检验;计数资料采用例和率表示,2组间比较采用χ2检验;2组细胞增殖曲线采用重复测量方差分析。P<0.05为差异有统计学意义。
2 结果
2.1 PRKG1在前列腺癌组织中的表达及其与临床分期和Gleason评分的关系 在PRKG1阳性表达方面,主要在细胞胞浆内观察到棕黄色染色,表明PRKG1高表达(见图1)。组织芯片免疫组化分析结果显示,与正常前列腺组织组[41.7%(5/12)]相比,PRKG1在前列腺癌组织[13.3%(10/75)]中阳性率显著降低(P=0.026),并且不同肿瘤临床分期和不同Gleason评分前列腺癌组织的PRKG1阳性率差异无统计学意义(P>0.05),见表1。
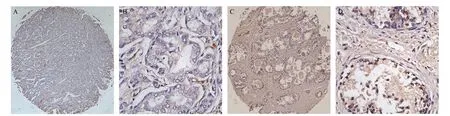
图1 PRKG1在前列腺癌组织和正常前列腺组织中的表达情况
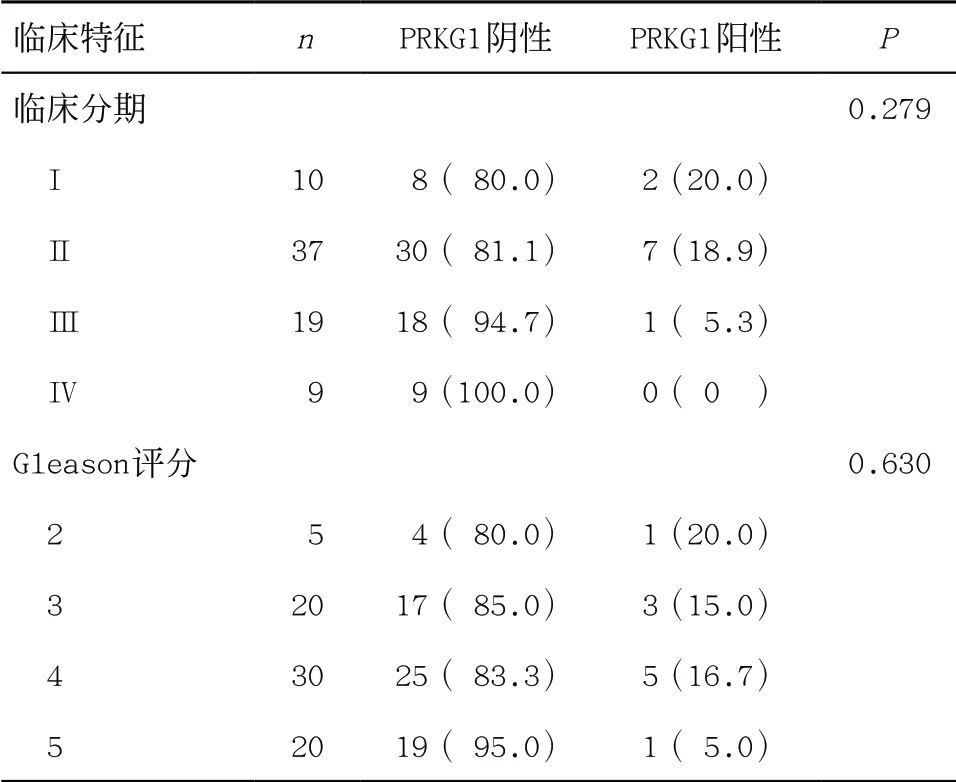
表1 前列腺癌组织PRKG1表达与临床分期及Gleason评分的关系[例(%)]
2.2 CRISPR/Cas9介导的PRKG1基因敲除促进细胞增殖、迁移和侵袭 Western blot结果显示,22RV1-KO组细胞不表达PRKG1,表明22RV1-KO组细胞中PRKG1基因敲除成功(见图2)。细胞增殖实验表明,与22RV1-CON组细胞相比,22RV1-KO组细胞的增殖水平增高,差异有统计学意义(P<0.05),见图3。Transwell试验结果进一步证实,与22RV1-CON组细胞相比,22RV1-KO组细胞迁移及侵袭能力均加强,差异有统计学意义(P<0.05),见图4。
2.3 PRKG1基因敲除对细胞凋亡蛋白、MMP9、细胞黏附蛋白及细胞骨架连接蛋白表达水平的影响 与22RV1-CON组细胞相比,22RV1-KO组细胞MMP9、CDH-1、Bax蛋白相对表达量降低(P<0.05),CDH-2、Bcl-2蛋白相对表达量升高(P<0.05),而Ezrin蛋白表达量差异无统计学意义(P>0.05),见图5。
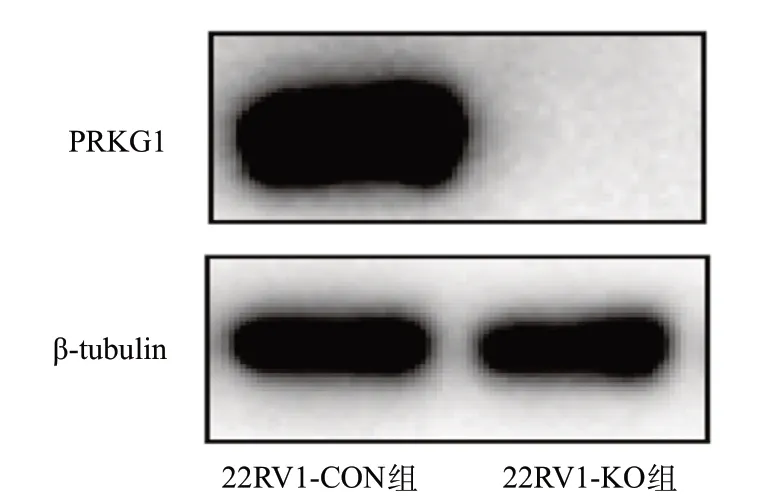
图2 Western blot确认22RV1细胞的PRKG1基因敲除结果
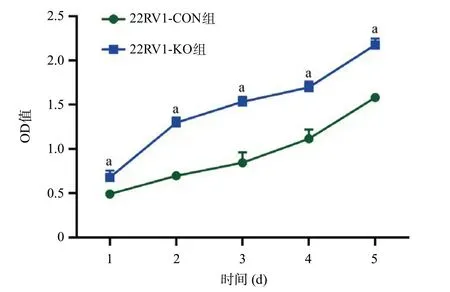
图3 PRKG1基因敲除对22RV1细胞增殖的影响
3 讨论
PRKG是广泛存在于真核生物中的丝氨酸/苏氨酸激酶。在哺乳动物细胞中发现了两种主要形式的PRKG:PRKG1和PRKG2[5]。过去的研究证明,与正常组织相比,部分肿瘤组织中PRKG1水平降低,且PRKG1通过蛋白的磷酸化影响细胞的活动[6]。人类集成蛋白质表达数据库(the Human Integrated Protein Expression Database,HIPED)提示PRKG在外周血单核细胞和前列腺正常组织中过表达。越来越多的证据表明,PRKG是可能诱导癌细胞凋亡的一种新的分子途径[7-8]。
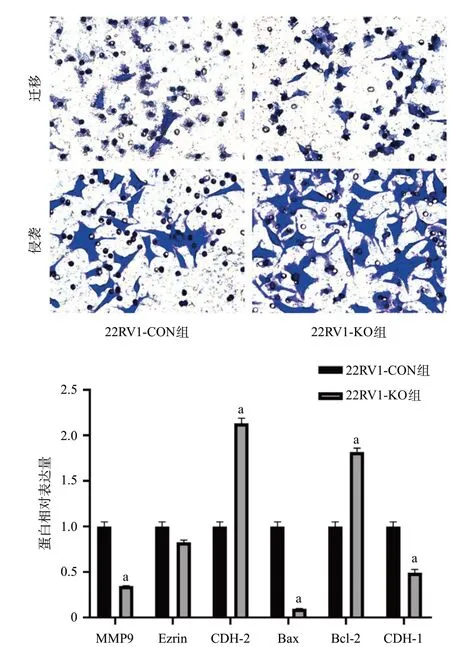
图5 2组细胞MMP9、Ezrin、CDH-2、Bax、Bcl-2、CDH-1蛋白相对表达量比较
另外有研究表明,某些抑癌基因,例如TCF12可通过cGMP-PRKG通路抑制前列腺癌细胞的增殖、迁移和侵袭[9]。但也有研究表明,TMPRSS2-ERG基因融合可导致ERG致癌通路异常激活,强化前列腺癌细胞cGMP合成,导致PRKG活性增强引起细胞增殖[10]。
前列腺癌发病机制是多种遗传因素共同参与的[11]。有研究表明PRKG1是前列腺癌进展过程中的一个抑癌基因[9]。我们观察到在前列腺癌组织中PRKG1较正常前列腺组织低表达,这些结果与PRKG1的抗增殖和抗肿瘤作用是一致的[12]。磷酸化是一种表观遗传学的机制,广泛参与肿瘤的发生发展[7,13],部分通路激活PRKG1可诱导前列腺癌的细胞凋亡,抑制前列腺癌细胞迁移[9,14]。舒林酸及其衍生物可能通过激活PRKG诱导细胞凋亡以预防结直肠癌、前列腺癌和乳腺癌[15-16]。
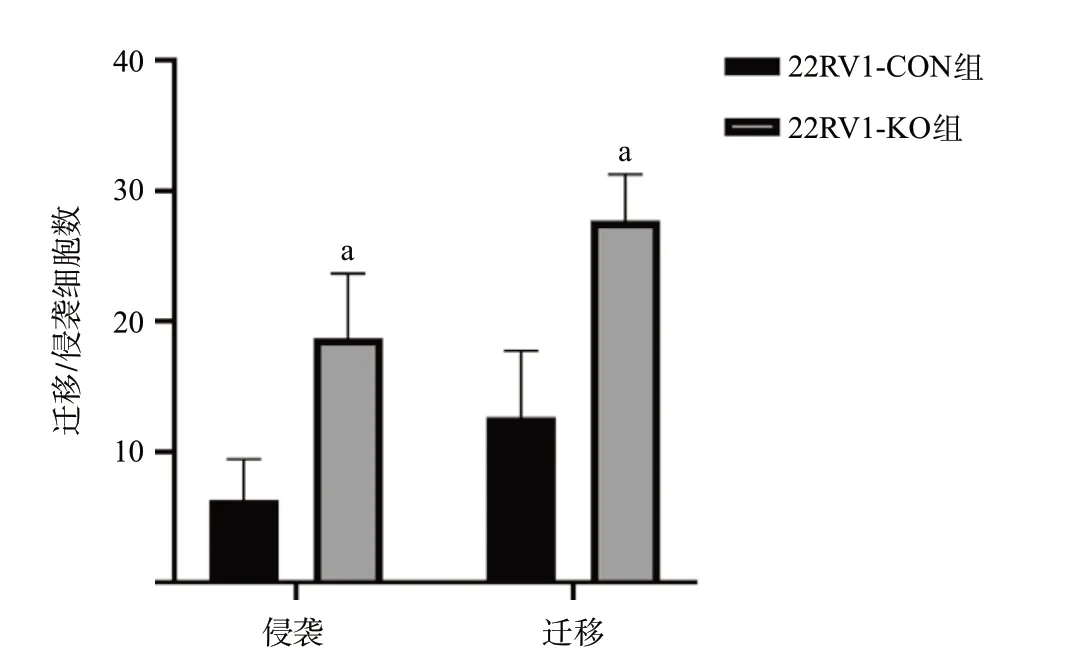
图4 PRKG1基因敲除对22RV1细胞迁移及侵袭能力的影响(×400)
本研究采用CRISPR/Cas9方法敲除PRKG1基因,发现在22RV1前列腺癌细胞中PRKG1基因的敲除降低了PRKG1的表达,同时敲除PRKG1基因后肿瘤细胞增殖、迁移及侵袭能力均上调,Western blot实验也发现了部分凋亡蛋白在22RV1-KO细胞系中表达的异质性。MMP9、CDH-1、Bax蛋白在22RV1-KO细胞克隆中表达下降,CDH-2、Bcl-2水平升高。MMP9虽是促癌基因,但在22RV-1中表达降低,这可能是由于cGMP激活中由NO模仿介导的炎症反应中,在基因水平上也降低了MMP9造成[17]。CDH-1、Bax抑制癌细胞增殖和迁移[18-19],在本研究中22RV1-KO细胞克隆敲除后CDH-1、Bax表达下降,验证了PRKG1是抑制前列腺癌生长与转移的。有研究报道,CDH-1可抑制恶性肿瘤发展,与BRAF、Src、AKT等多条癌症相关信号通路存在相互作用,靶向CDH-1策略已成为当前肿瘤防治研究突破新方向[20]。CDH-2是一种促进恶性肿瘤的基因[21],CDH2介导的细胞黏附是生存信号的重要中介,并抑制了细胞的凋亡[22]。本研究PRKG1敲除后,同时通过下调上皮细胞标志物CDH-1与上调间质标志物CDH-2来影响前列腺癌细胞的迁移与侵袭能力。已有研究发现有关细胞凋亡基因分为以Bax为代表的促进细胞凋亡的基因和以Bcl-2为代表一致细胞凋亡的基因[23]。我们的结果中凋亡蛋白Bcl-2抑制和促进凋亡蛋白Bax升高,与我们的研究预期相符。促癌基因Ezrin比值变化差异无统计学意义,可能与PRKG1基因无明显相关性。
本研究结果证明了PRKG1的重要性,并为其作为前列腺癌治疗药物靶点提供了基础。但研究还有很大的局限性。例如,组织芯片中并没有患者生存期信息,研究敲除PRKG1影响的蛋白种类有限,未研究PRKG1过表达对前列腺癌细胞的影响,缺少动物实验等。我们认为,这个基因还需要更深入的研究评估,以便更好地理解其治疗潜力。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
昆明医科大学学报(2022年1期)2022-02-28
基础医学与临床(2022年1期)2022-01-21
临床与实验病理学杂志(2021年7期)2021-12-03
医学食疗与健康(2021年27期)2021-05-13
昆明医科大学学报(2021年1期)2021-02-07
现代临床医学(2021年1期)2021-01-26
学校教育研究(2018年27期)2018-05-14
晚晴(2017年11期)2017-11-12
中国民族民间医药·下半月(2016年8期)2016-10-24
