
Genomic selection:A breakthrough technology in rice breeding
2021-06-19 07:36:34YngXuKexinYueZhoXinWngKiZhouGungningYuChengLiPengchengLiZefengYngChenwuXuShizhongXu
The Crop Journal 2021年3期
Yng Xu ,Kexin M ,Yue Zho ,Xin Wng ,Ki Zhou ,Gungning Yu ,Cheng Li ,Pengcheng Li ,Zefeng Yng ,Chenwu Xu ,,Shizhong Xu
a Key Laboratory of Plant Functional Genomics of the Ministry of Education/Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding/Jiangsu Co-Innovation Center for Modern Production Technology of Grain Crops,College of Agriculture,Yangzhou University,Yangzhou 225009,Jiangsu,China
b Department of Botany and Plant Sciences,University of California,Riverside,CA 92507,USA
Keywords:Genomic selection Rice Hybrid Predictive ability Model
ABSTRACT Rice(Oryza sativa)provides a staple food source for more than half the world population.However,the current pace of rice breeding in yield growth is insufficient to meet the food demand of the everincreasing global population.Genomic selection(GS)holds a great potential to accelerate breeding progress and is cost-effective via early selection before phenotypes are measured.Previous simulation and experimental studies have demonstrated the usefulness of GS in rice breeding.However,several affecting factors and limitations require careful consideration when performing GS.In this review,we summarize the major genetics and statistical factors affecting predictive performance as well as current progress in the application of GS to rice breeding.We also highlight effective strategies to increase the predictive ability of various models,including GS models incorporating functional markers,genotype by environment interactions,multiple traits,selection index,and multiple omic data.Finally,we envision that integrating GS with other advanced breeding technologies such as unmanned aerial vehicles and open-source breeding platforms will further improve the efficiency and reduce the cost of breeding.
1.Introduction
The primary mission of rice breeding is to develop cultivars with high yield,nutritious,pest-and disease-resistant,and climate-smart[1].Conventional breeding based on hybridization of parents and phenotypic selection of offspring is timeconsuming.It takes~10 years to develop and release a novel rice variety.Since the 1990s,advances in molecular technology allow breeders to use DNA markers to assist selections.Marker-assisted selection(MAS)was proposed to select individuals with QTL-associated markers.Although MAS can shorten breeding time,it is less suitable for quantitative traits influenced by many genes with small effects[2].Genomic selection(GS)has been proposed as a promising tool to overcome the limitation[3].GS uses genome-wide DNA markers and phenotypes of target traits from a training population to predict genomic estimated breeding values(GEBVs)of candidates in a test population,where the latter have been genotyped but not yet phenotyped[4].The advantages of GS over traditional MAS include that GS does not need to detect significant QTL related to traits of interest and it greatly increases the efficiency of breeding due to early selection before the phenotypic data are collected.Motivated by the huge success in improving the genetic gain of animal breeding,GS has been introduced to crop breeding in many aspects such as inbred performance prediction,parental selection,and hybrid prediction[5-8].When applied to hybrid breeding,GS is more effective because genotypes of hybrids can be deduced from genotypes of their parents,rather than sequenced anew,substantially reducing the sequenced cost[9].Recently,the usefulness of GS in rice breeding has been proved by several simulations and experimental studies[10-14].
High prediction accuracy is a prerequisite for successful application of GS.The prediction accuracy is often measured by the correlation between the observed phenotypes and the predicted GEBVs or predicted phenotypes(predictive ability)using k-fold cross-validation(CV),where the population is randomly partitioned into k equal sized parts and each part is predicted once using parameters estimated from the other k-1 parts[15].The predictive ability is influenced by several factors,including population size,relatedness among individuals within the training population and between the training and the test populations,heritability of traits,marker density,statistical model and so on[4,10].According to the breeder’s equation,even a small increase in predictive ability can be converted into a huge gain with a strong selection intensity.Several strategies have been attempted to increase the predictive ability for complex traits with low heritability such as grain yield[16].In this review,we aim to give a brief introduction to key factors affecting GS,summarize current progress on the application of GS in rice,emphasize the strategies to increase the predictive ability,and discuss some perspectives for future development of GS in crop breeding.
2.Key factors affecting GS
2.1.Genetic factors affecting the predictive ability
When applying GS to crop breeding,several genetic factors should be carefully considered,including marker density,sample size,the relationship between the training population and test population,population structure,heritability and genetic architecture of target traits,and linkage disequilibrium(LD)between markers and QTL.In general,the predictive ability grows as marker intensity and sample size grow until reaching a plateau.GS based on a population of 363 elite rice breeding lines revealed that there was no significant difference in the predictive ability when using 7142 SNPs or 73,147 SNPs[11].Another GS experiment for 575 rice hybrids genotyped with 2,054,293 SNPs indicated that the predictive ability reached a plateau at 5K SNPs[10].The necessary size of the training population is associated with the heritability of the target trait and population relatedness.For low heritability traits(e.g.,h2=0.2),more than 1000 individuals are required in the training population[17].Additionally,the desirable size of training population is much smaller for a closely related population than that for a distantly related population.By using three methods of representative subset selection,Guo et al.[18]demonstrated that effective GS models can be built with a training population 2%-13%of the whole population.The genetic relationship between the training and the test populations is an important factor affecting the predictive ability,and more accurate prediction can be achieved for genetically similar populations[19,20].A prevalent view is that higher predictive ability can be obtained by adding more related materials in the training population rather than increasing the size of the training population with unrelated materials.However,increasing relatedness will damage the genetic gain in the long run as the genetic variation will be limited or exhausted if the related populations are overused.Therefore,the relationship between tanning population and test population should be balanced and optimized in practical breeding[21].Population structure influences performance of genomic prediction in stratified populations,which leads to spurious associations between markers and traits,thus giving biased effect estimates and predictive ability[22,23].The degree of LD between markers and QTL also affects GS.To maintain the predictive ability over time,the training population needs to be updated regularly because the LD between markers and QTL will gradually decrease as the number of generations grows[2].The predictive ability is closely related to the heritability of traits.Jia et al.[24]demonstrated that the heritability calculated through cross validation is equivalent to trait predictive ability.High heritability traits such as plant height often have higher predictive abilities than low heritability traits such as grain yield.
2.2.Statistical models for GS
In addition to the above genetic factors,the statistical method is another important factor affecting the predictive ability.A large number of parametric and non-parametric statistical methods have been used for GS.Parametric methods mainly include genomic best linear unbiased prediction(GBLUP),ridge regression best linear unbiased prediction(RRBLUP),partial least squares(PLS),least absolute shrinkage selection operator(LASSO),elastic net,and Bayesian methods(BayesA,BayesB,BayesC,BayesCπ,BayesR and Bayesian LASSO,etc.);non-parametric methods include random forest(RF),support vector machine(SVM),reproducing kernel Hilbert space(RKHS),deep learning and so on.The GBLUP method assumes all marker following the same genetic variance and uses the genomic relationship matrix to predict phenotypes without estimating marker effects[25].Hence,the GBLUP method is robust and fast,and more suitable for highly polygenic traits.RRBLUP has been shown to be mathematically equivalent to GBLUP[26].Variable selection methods such as LASSO,elastic net and ridge regression shrink the effects of most loci towards zero,which fit data well for traits controlled by major genes[25,27].The main feature of Bayesian methods is that allow different markers to follow specific prior distributions[28].For instance,BayesB assumes a two-component mixture prior distribution with one component being a point mass at 0 and the other following a scaled-t distribution[3].BayesR assumes a mixture of four normal distributions mixture and one component of the mixture with zero variance,accommodating markers with major,moderate,minor,or no effect[29].The unknown parameters of Bayesian methods are usually estimated using the Markov Chain Monte Carlo(MCMC)algorithm,resulting in a high computational burden.Simulation studies revealed that the Bayesian methods are sensitive to the genetic architecture of traits and they are better for traits with large effects[30].The SVM and RKHS methods are kernel-based supervised learning method for classification and regression,which are more effective for capturing non additive effects[31,32].The RF method is to build a large number of trees called forest by introducing two layers of randomness,one is the random bootstrap sampling of data,the other is the random selection of a subset of predictors at each node[33].This method takes the average of all tree nodes to find the best prediction model.Deep learning is a multi-layer perceptron with multiple hidden layers,which enable capture of the complex non-linear relationships contained in the data.However,deep learning needs huge dataset to make accurate prediction possible[34,35].Many researchers have compared the predictive ability of these methods using simulation and real data[36].However,no method fits all the data universally optimal.Xu et al.[37]suggested that treating GS method as a parameter and evaluate the predictive abilities of all available methods using cross-validation,and then select the best method with the maximum accuracy in the corresponding GS program.For convenience,the software commonly used in GS are summarized in Table 1.
3.GS application in rice
3.1.GS application in pure line selection
GS technology can be used for inbred selection as well as hybrid breeding in rice.Currently,GS studies in rice mainly focus on designing a training population and evaluating the predictive ability within or between populations.In rice breeding populations,genomic prediction has been performed for various quantitative traits and moderate to high predictive ability has been reported(Table 2).These studies show the feasibility and great potential of GS in rice breeding pipelines.Onogi et al.[38]obtained average predictive ability of heading date(~0.8),culm length(~0.8),panicle length(~0.6),panicle number(~0.4),grain length(~0.4),and grain width(~0.6)for a population of 110 Asian rice cultivars using nine prediction methods.Based on a diverse population of 413 rice inbred lines from 82 countries genotyped with a 44 K chip,Isidro et al.[39]compared five different sampling strategies and found that stratified sampling showed the highest predictive abilitiesfor florets per panicle,flowering time,plant height,protein content with~0.6,~0.63,~0.7,and~0.44,respectively.Iwata et al.[40]reported a high predictive ability of~0.6 for rice grain shape based on 386 rice diverse accessions.Spindel et al.[11]performed GS on a panel of 363 elite breeding lines from the International Rice Research Institute’s(IRRI)irrigated rice breeding program and reported predictive abilities of 0.31,0.34,and 0.63 for grain yield,plant height and flowering time,respectively.For 343 S2:4lines from an upland rice synthetic population,the highest predictive abilities were obtained for grain yield(0.309)with Bayesian LASSO,plant height(0.538)with Bayesian ridge regression,and flowering time(0.295)with LASSO[41].Based on a training population of 284 inbred lines,Hassen et al.[42]assessed the predictive ability for the progeny of biparental crosses comprising 97 F5-F7lines from 36 biparental crosses and found that the average predictive abilities of progeny predictions were 0.35 for flowering date,0.33 for nitrogen balance index,and 0.38 for100 panicle weight,which were slightly lower than the predictive ability obtained from the cross validation in the training population.Yabe et al.[12]developed an approach for describing grain weight distribution using 128 Japanese rice varieties and performed GS for Grain-filling characteristics.The proportion of filled grains and the average weight of filled grains,important traits related to grain weight distribution,were predicted with predictive abilities of 0.30 and 0.28,respectively.In addition to agronomic traits,Huang et al.[43]also proposed that GS could be useful for predicting rice blast,with predictive abilities of the GBLUP model ranging from 0.15 to 0.72 across isolates.
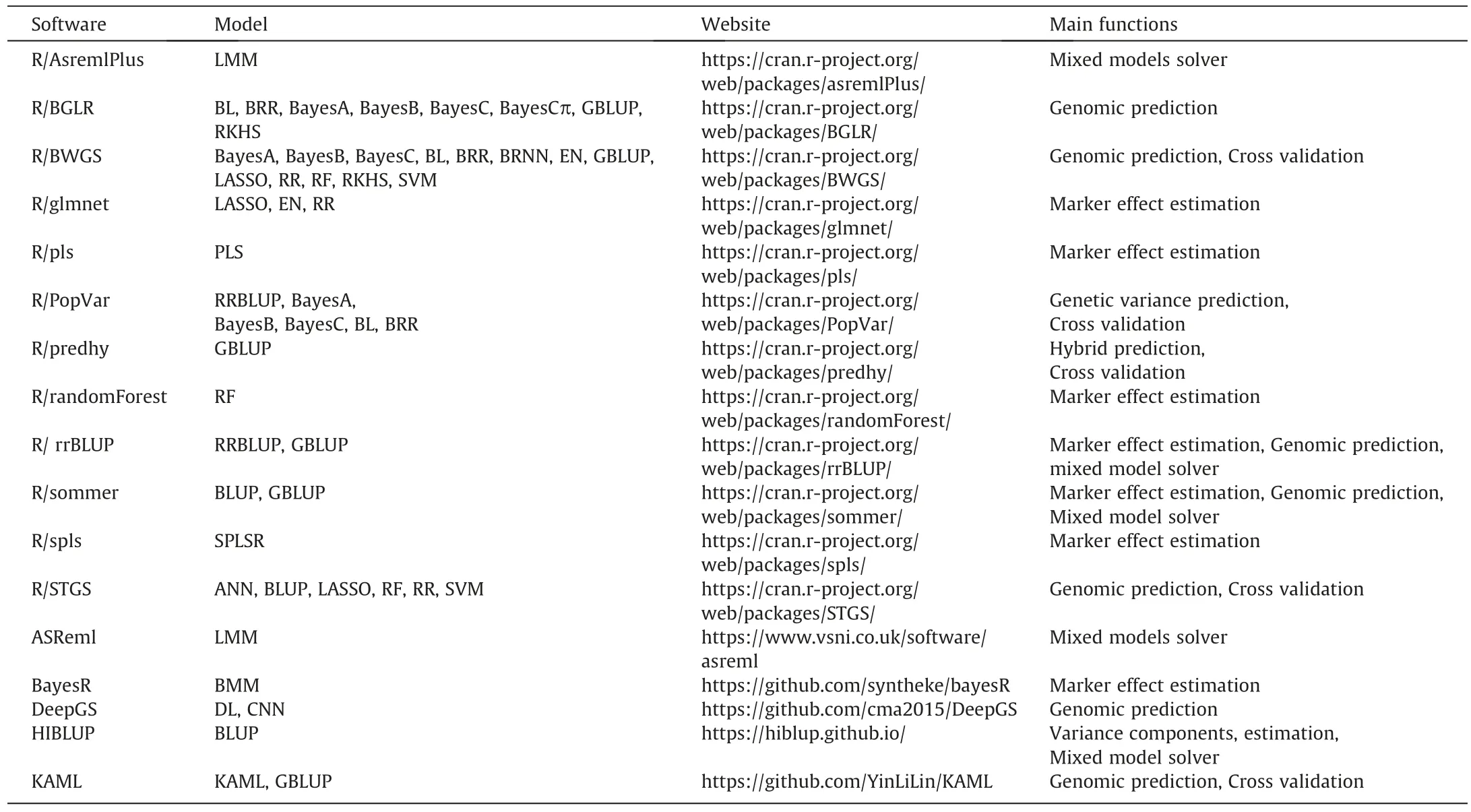
Table 1Commonly used software for genomic selection in plants.
3.2.GS application in hybrid breeding
Hybrid breeding is a main tool to increase grain yield in rice yield by taking advantage of heterosis.Hybrid rice has a 20%yield advantage over inbred varieties[44].The biggest challenge in rice hybrid breeding lies in selecting desired hybrids out of numerous potential crosses.It is unrealistic to assess the field performance of all potential hybrids due to limited resources.Fortunately,GS has paved the way to solve the problem.In hybrid rice breeding,GS predicts the performance of all combinations of a given set of genotyped parents with only a small proportion of all potential crosses required to be evaluated in the field,which substantially saves the cost of hybridization and field experiments[10].For the first time,Xu et al.[9]predicted hybrid performance of rice using the GBLUP method with a marker inferred kinship matrix rather than pedigree information of the hybrids.They randomly paired 278 crosses from 210 recombinant inbred lines derived from a cross between Zhenshan 97 and Minghui 63 as a training population and predicted the remaining 21,667 untested hybrids.The predictive abilities of yield and 1000 grain weight drawn from five-fold cross validation are 0.36 and 0.82,respectively.They predicted that selection of the top 100 crosses would lead to a 16%increase in grain yield compared with all 21,945 potential crosses.This study offers a proof of concept for hybrid prediction in rice.For general application to a broader range of germplasms,experimental designs involving a subset of the crosses must be used.Wang et al.[7]predicted hybrid performance in rice using North Carolina mating design II(NC II).Using 575 rice hybrids generated by crossing 115 inbred lines with five male sterile lines as training popula-tion,they predicted the performance of 6555 potential crosses between the 115 inbred lines and found that the average predicted grain yield of the top 100 crosses(51.78)was much higher than that of all potential hybrids(38.94).The study demonstrated the usefulness of NC II design for hybrid prediction in rice.Some publicly available germplasms and data provide a great opportunity for rice hybrid breeding.For example,the 3000 rice genomes project(3 K RGP)sequenced over 3000 diverse accessions collected from 89 countries[45].A total of 29 million SNPs were released by the 3 K RGP.Based on the same training population of 575 hybrids,Xu et al.[10]predicted the performance of 362,760 potential crosses between the 120 rice lines and the 3023 rice accessions from 3 K RGP and concluded that average grain yield of the top 100 predicted crosses represented a 35.5%increase compared with that of all potential crosses.Recently,Cui et al.[46]proposed a novel strategy that used the existing hybrids as training population to predict hybrids from seemingly unrelated parents,which enabled selection of superior hybrids with the minimum cost.To validate this strategy,they used an existing population of 1495 rice hybrids as training population to predict 100 hybrids derived from half diallel crosses involving 21 parents which were not included in the parents of the training population.The relatively high predictive abilities for six traits were reported,which demonstrated the viability of the proposed strategy.They also used this training population to predict 44,636 potential hybrids in a three-line mating system from the 3 K RGP and selected 200 predicted hybrids based on selection index of all traits.To visually demonstrate the results of hybrid prediction of the above studies,boxplots were drawn for grain yield of the top 200 and bottom 200 crosses out of 21,945,6555,362,760,and 44,636 potential crosses predicted by Xu et al.[9],Wang et al.[7],Xu et al.[10]and Cui et al.[46],respectively(Fig.1a-d).The corresponding average grain yield of the top 200 selection is 50.1,50.4,51.0 and 40.6,while that of the bottom selection is 36.9,24.1,19.8,and 33.4,respectively.It can be found that the grain yield of the top 200 crosses predicted from different populations have similar predicted values,suggesting the reliability of GS in rice breeding.There are significant differences in the predictive abilities between the top 200 crosses and the bottom 200 crosses.These top and bottom predicted crosses can be further validated in the field.

Table 2Summary of genomic selection studies in rice.
However,there are still limited studies applying GS to rice breeding practice,especially when compared to other major crops such as maize and wheat.The International Maize and Wheat Improvement Center(CIMMYT)have implemented GS in global maize breeding program[47].For example,Zhang et al.[48]designed a rapid cycling genomic selection(RCGS)of multiparental crosses.Eighteen elite tropical maize lines were intercrossed twice and self-pollinated once to form the cycle 0(C0),which was genotyped and phenotyped at four locations in Mexico.Individuals with the best phenotype were selected to form the parents for RCGS cycle 1(C1).Predictions of the genotyped individuals forming C1 were performed,and the best predicted lines were selected as parents of C2.This procedure was repeated for more cycles(C2,C3,and C4)and two cycles per year were achieved.The realized grain yield from C1 to C4 reached 0.225 t ha-1per cycle,which is equal to 0.1 t ha-1per year over a 4.5-year breeding period,demonstrating GS is an effective breeding strategy for conserving genetic diversity and achieving high genetic gains in a short period simultaneously.

Fig.1.Comparison of the top 200 hybrids and the bottom 200 hybrids out of all potential hybrids for predicted grain yield.(a)The top 200 hybrids and the bottom 200 hybrids out of all 21,945 hybrids predicted by Xu et al.[9].(b)The top 200 hybrids and the bottom 200 hybrids out of all 6555 hybrids predicted by Wang et al.[7].(c)The top 200 hybrids and the bottom 200 hybrids out of all 362,760 hybrids predicted by Xu et al.[10].(d)The top 200 hybrids and the bottom 200 hybrids out of all 44,636 hybrids predicted by Cui et al.[46].
4.Strategies for increasing predictive ability
4.1.Incorporating markers into the GS model

Fig.2.Main strategies for increasing the predictive ability.
Several strategies have been attempted to increase the predictive ability for complex traits(Fig.2).Incorporating prior information about known genes or identified SNPs in GS model potentially reveals the genetic architecture of complex traits and is expected to improve predictive ability.Based on a simulation study,Bernardo et al.[49]suggested that when a few major genes are known and each major gene explains larger than 10%of genetic variance,these major genes should be fitted as fixed effects rather than random effects in the BLUP model to improve the prediction.In the absence of prior knowledge of genes,significant or peak SNPs identified by GWAS can also be treated as fixed effect covariates.Zhang et al.[50]demonstrated that incorporating the peak SNPs from publicly available GWAS results as fixed effects improved the prediction for most traits in a rice diversity panel.Spindel et al.[51]proposed an approach called GS+de novo GWAS,which incorporated markers as fixed effects from the results of GWAS conducted on the same training population of GS.In a IRRI rice breeding population,the proposed method outperformed six other methods across all traits and led to a 12.8%increase for grain yield in predictive ability compared with the model without the fixed effects.Notably,the efficiency of the joint strategy combing GWAS and GS largely relies on the genetic architecture of given traits.Bian et al.[52]and Rice et al.[53]found that the joint strategy was more suitable for traits with a few large effect QTNs in a polygenic background.Therefore,the genetic architecture of target traits shouldbe investigated before applying this strategy to a breeding program.
4.2.Incorporating genotype by environment interactions into the GS model
Multi-environment trials have been routinely conducted in crop breeding.Incorporating genotype by environment(G×E)interaction allows borrowing information between correlated environments[54].Several studies have developed GS models that accommodate G×E interaction and reported that these models result in considerable gain in predictive ability compared to the single environment counterparts.López-Cruz et al.[55]extended the GBLUP method that incorporated G×E interactions by modeling interactions between all markers and environments.The main feature of their model is that it can separate markers having common effects across environments and markers having environment-specific effects.In breeding programs,this may facilitate selection of candidates with adaptability and stability.Cuevas et al.[56]further incorporated nonlinear Gaussian kernels with the G×E model of López-Cruz et al and found that the predictive ability of their model increased up to 17%based on a CIMMYT wheat dataset.Bayesian models including Bayesian ridge regression and BayesB have also been extended for dealing with G×E interaction[57,58].The G×E models in rice breeding are advantageous over the single environment GS models.Based on a rice reference population and a progeny population assessed under two water management systems,Hassen et al.[59]first investigated the effectiveness of breeding for adaptation to specific abiotic stress using GS models including G×E interaction and found that the predictive ability of G×E models for predicting unobserved phenotypes under two environments increased by an average of~30%over single environment models.In a training population of 280 IRRI breeding lines evaluated under a favorable and two managed drought environments,Bhandari et al.[13]demonstrated the RKHS model incorporating G×E interaction achieved predictive ability for tolerance to drought stress up to 32%higher than single environment GS models when the test population has been evaluated under at least one environment.
4.3.Multi-trait GS methods
In breeding practice,multiple traits should be considered simultaneously for selection.Multi-trait GS models have been shown to be beneficial for increasing predictive ability for traits with low heritability.Genetic and residual correlations among traits provide additional information for genomic selection and thus improve predictive ability[60].Some of the single-trait GS models have been extended to multiple trait selection,such as the multivariate GBLUP and multivariate Bayesian methods[61,62].The initial multivariate models assumed that each locus simultaneously affects multiple traits or none of them.Cheng et al.[63]proposed a general multi-trait BayesCπand BayesB method,allowing each locus to influence any combination of multiple traits rather than all of them.They developed an open-source software JWAS to perform multi-trait GS.Computation complexity is the main limitation for multi-trait GS.Recently,Wang et al.[60]established a bivariate GS(2D GS)model by incorporating the HAT methodology to BLUP model,which substantially increases the computational efficiency by avoiding cross-validation.Base on a rice dataset of 210 RILs,they confirmed that the predictive abilities for any two traits achieved from 2D GS analysis were higher than those obtained from the single-trait analysis.
Multi-trait GS models can also be used to predict traits of interest that are difficult or costly to evaluate such as root traits and grain yield per plot using auxiliary traits that are convenient to measure such as plant height.Wang et al.[7]developed a multivariate GBLUP model using a multivariate kinship matrix constructed by auxiliary variates.They used the proposed method to predict rice hybrid performance and concluded that the average predictive ability of the multivariate GBLUP with two auxiliary traits was 6.4%higher than that of the univariate GBLUP,while the gains of the multivariate GBLUP with eight auxiliary traits over the univariate GBLUP is up to 26.7%.Sun et al.[64]demonstrated that incorporating canopy temperature and vegetation index measured from high-throughput phenotyping(HTP)platforms as the auxiliary traits for grain yield in a multivariate GS model increased the predictive ability by up to 70%compared with the univariate model.
In addition to the multivariate GS model,selection index can be used for breeding selection of multiple traits simultaneously.In the context of GS,genomic selection index,a linear combination of GEBV of the traits weighted by respective economic values,has been constructed for predicting the net genetic merit and selecting parents[65,66].Recently,Wang et al.[67]developed a selection index-assisted GS method that takes advantage of the feature of selection index to aggregate information of auxiliary traits in the training population to predict target traits with low heritability in the test population.They evaluated the predictive ability of this method in simulated and real hybrid rice dataset and concluded that the selection index-assisted GS is significantly superior over the single-trait GS model.
4.4.Integrating multi-omic data for hybrid prediction
Several studies demonstrated that the predictive ability is often low for some complex traits,especially for yield traits greatly affected by environment.Typical genomic selection methods are not sufficient to capture interactions between genes and their downstream regulators[68].Downstream‘‘omes”including transcriptome,proteomes and metabolome reflect interactions within and between various biological strata[69].As advancement of omics technologies,metabolomic and transcriptomic data offer novel sources for phenotypic prediction in several crop species.Some researchers have attempted to use parental transcriptomic or metabolomic data to predict the performance of unobserved hybrids.
With respect to transcriptomic prediction,Frisch et al.[70]for the first time used the expression profile data of 21 parental inbred lines and phenotypic data of 98 hybrids to perform GS for maize hybrid performance.Based on the same dataset,Fu et al.[71]used four methods including multiple linear regression,PLS,SVM,and transcriptome distance to predict phenotypes of maize hybrids and found that prediction based on transcriptome distances was the most accurate method.Zenke-Philippi et al.[72,73]used 46 k microarray chip data,2 k core gene expression data,and 1 k AFLP marker data to perform genomic and transcriptomic prediction on yield and dry matter content of maize hybrids.Under the ridge regression model,the transcriptomic prediction of the hybrid performance was slightly better than the genomic prediction.
For metabolomic prediction,Meyer et al.[74]first used metabolites to predict biomass in Arabidopsis,and the correlation between the predicted value and the true value reached 0.58.By crossing 285 maize inbred lines with two testers,Riedelsheimer et al.[5]predicted the combining abilities of 7 agronomic traits and found that the prediction accuracy of 130 metabolites was no less than that of 56,110 SNP markers.Xu et al.[75]used the metabolic data of 210 rice parents to predict the yield of hybrids and found that the predictive ability was almost doubled compared to genomic prediction.The predicted yield of the top 10 hybrids based on metabolites was about 30%higher than the average predicted yield.Based on metabolic profile data of 18 rice parents andthe yield-related traits of 306 hybrids derived from their diallel crosses,Dan et al.[76,77]reported high predictive abilities of metabolomic prediction using the PLS method.The sum,difference and ratio of parental metabolite level were used to construct the prediction models,respectively.
To make full use of omic information,the joint prediction research of multi-omic data has attracted much attention from plant breeders.Westhues et al.[69]revealed the superiority of integrating transcriptomic data with genomic data measured on parent lines for hybrid prediction.Wang et al.[78]compared the predictive abilities of all combinations of three-omic data with eight GS methods and concluded that the predictive performance obtained from the combination of genomic and metabolic data with the GBLUP method was overall the best for selecting hybrid rice.
Although the above studies demonstrated that both transcriptome and metabolome are effective predictors for predicting hybrids,there are still some issues that need to be further explored.Prediction based on transcriptomic and metabolomic information should select the suitable tissue and sampling time due to the dynamic nature of transcript and metabolite profiles[79].Additionally,current transcriptomic and metabolomic data of hybrids usually adopt the same coding method of genomic data.It is inevitably biased because neither the transcriptome nor the metabolome can be inferred from the omic information of their parents directly like the genome.The quantitative relationship between transcript and metabolite levels of hybrids and those of their parents needs to be further investigated.In breeding practice,the trade-off between improvement of the predictive ability and increase in the cost need to be considered.In addition to parental transcriptomic and metabolomic data,parental phenotypic data can also be used to predict hybrids.Recently,Xu et al.[16]proposed a novel strategy of incorporating parental phenotypes into multi-omic prediction models for predicting rice hybrids.They demonstrated that integrating parental phenotypes with any other omic predictors has significantly increased the predictive ability for yield-related traits in rice.Incorporation of parental trait values does not require additional cost because parental traits are always available.
5.Perspectives
To further accelerate the breeding process and reduce the breeding cost,we should combine GS with other advanced breeding technologies and platforms.HTP platforms enable acquisition of large-scale phenotypic data in controlled environments and fields with high accuracy and low labor intensity[80].HTP combined with other strategies can improve heritability estimation and prediction accuracy.Recently,remote sensing with unmanned aerial vehicles(UAVs)provides novel opportunities in field phenotyping.UAVs offer a flexible platform and provide high quality hyperspectral data with centimeter resolution[81].The secondary traits such as vegetation index and 3D plant canopy structure obtained by UAVs platforms can be incorporated into GS models to improve the predictive ability of target traits and genetic gain[82].To take full advantage of UAV information,further studies are warranted to define ideal sensor configuration and develop specific models and software.The GS technology equipped with UAV platforms is expected to become an effective and routine strategy in crop breeding.
In GS breeding,genotyping generally consumes substantial amounts of breeding cost.Nowadays,genotyping-by-sequencing(GBS)technology has been widely used to obtain high density SNPs in GS studies,but it requires bioinformatic analysis procedure and heavy imputation as well as difficulty in data sharing[83].In contrast,SNP arrays are designed based on standardized procedures and fixed loci,which can be applied for quickly genotyping large samples and the data analysis is simple for breeders[84].SNP arrays including 44 K SNP array,RICE6K,RiceSNP50,C7AIR have been designed for rice molecular breeding.Recently,Zhang et al.[85]characterized the gene-CDS-haplotype(gcHap)diversity of 45,963 rice genes in 3010 rice accessions and found that haplotype-based GS had higher predictive ability than SNP-based GS because that relatively a few gcHaps represent functional importance of most rice genes.However,several major genes have not been fully integrated into the existing rice array.The SNP array specific for rice GS breeding needs to be developed for global rice community.
To optimize breeding efficiency,open-source platforms have also been suggested for GS breeding[86].In open-source platforms,researchers and breeders can share their genotypic and phenotypic data of training populations planted in various environments.Based on the existing population dataset,researchers can reinforce their own GS models and predict candidates in local environments more quickly and accurately.To jointly analyze or reuse these data,the common criteria and data format need to be defined[21].Additionally,there is an urgent need to develop novel statistical models such as machine learning to handle the explosion of agriculture data.With the optimized experimental design,precise HTP platforms,low-cost genotyping technologies and improved models,GS technology will further improve rice varieties under the joint efforts of global researchers.
CRediT authorship contribution statement
Chenwu XuandShizhong Xuconceived the manuscript;Yang Xuwrote the original draft;Kexin Ma,Yue Zhao,Xin Wang,andKai Zhoudrew figures and collected data for tables;Cheng Li,Pengcheng Li,andZefeng Yangrevised the manuscript.All authors read and approved the final manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China(31801028,32061143030,and 41801013),the National Key Technology Research and Development Program of China(2016YFD0100303),the Priority Academic Program Development of Jiangsu Higher Education Institutions,the Innovative Research Team of Ministry of Agriculture,and the Qing-Lan Project of Yangzhou University.

- The Crop Journal的其它文章
- Breeding by design for future rice:Genes and genome technologies
- Innovation and development of the third-generation hybrid rice technology
- Understanding the genetic basis of rice heterosis:Advances and prospects
- CRISPR/Cas systems:The link between functional genes and genetic improvement
- Target chromosome-segment substitution:A way to breeding by design in rice
- Breeding by selective introgression:Theory,practices,and lessons learned from rice