
Breeding by selective introgression:Theory,practices,and lessons learned from rice
2021-06-19 07:36:32FanZhangYingyaoShiJauharAliJianlongXuZhikangLi
The Crop Journal 2021年3期
Fan Zhang ,Yingyao Shi ,Jauhar Ali ,Jianlong Xu *,Zhikang Li *
a Institute of Crop Sciences,Chinese Academy of Agricultural Sciences,Beijing 100081,China
b College of Agronomy,Anhui Agricultural University,Hefei 230036,Anhui,China
c Rice Breeding Platform,International Rice Research Institute(IRRI),Los Baños,Laguna 4031,Philippines
d Shenzhen Branch,Guangdong Laboratory for Lingnan Modern Agriculture,Genome Analysis Laboratory of the Ministry of Agriculture,Agricultural Genomics Institute at Shenzhen,Chinese Academy of Agricultural Sciences,Shenzhen 518120,Guangdong,China
Keywords:Backcross Trait-specific introgression line Breeding by selective introgression Designed QTL pyramiding
ABSTRACT Future demands for increased productivity and resilience to abiotic/biotic stresses of major crops require new technologies of breeding by design(BBD)built on massive information from functional and population genomics research.A novel strategy of breeding by selective introgression(BBSI)has been proposed and practiced for simultaneous improvement,genetic dissection and allele mining of complex traits to realize BBD.BBSI has three phases:a)developing large numbers of trait-specific introgression lines(ILs)using backcross breeding in elite genetic backgrounds as the material platform of BBD;b)efficiently identifying genes or quantitative trait loci(QTL)and mining desirable alleles affecting different target traits from diverse donors as the information platform of BBD;and c)developing superior cultivars by BBD using designed QTL pyramiding or marker-assisted recurrent selection.Phase(a)has been implemented massively in rice by many Chinese research institutions and IRRI,resulting in the development of many new green super rice cultivars plus large numbers of ILs in 30+elite genetic backgrounds.Phase(b)has been demonstrated in a series of proof-of-concept studies of high-efficiency genetic dissection of rice yield and tolerance to abiotic stresses using ILs and DNA markers.Phase(c)has also been implemented by designed QTL pyramiding,resulting in a prototype of BBD in several successful cases.The BBSI strategy can be easily extended for simultaneous trait improvement,efficient gene and QTL discovery and allele mining of complex traits using advanced breeding lines from crosses between a common‘‘backbone”parent and a set of elite parents in conventional pedigree breeding programs.BBSI can be relatively easily adopted by breeding programs with small budgets,but the BBSI-based BBD strategy can be fully and more efficiently implemented by large seed companies with sufficient capacity.
1.Introduction
Since the rediscovery of Mendelian genetic principles,the conventional plant breeding strategy of hybridization and phenotypic selection has been practiced for more than a century and has led to the improved productivity of crops today.Backcross(BC)breeding is used for transferring a highly heritable trait into an elite cultivar[1].Progeny in a BC population are phenotypically similar to their recurrent parent(RP)but carry one or several target trait(s)introgressed from a known donor.Conventional BC breeding requires backcrossing and screening of target traits in each BC generation.In the BC breeding process,the donor genome is randomly introgressed into the RP background and the proportion of the RP genome in BC progeny is recovered at a rate of 1-(1/2)t+1for each BC generation(where t is the number of backcrosses),though plants of a specific BC population vary widely in the amount of donor genome they carry[2].Thus,only plants with the highest proportion of RP genome and the target trait are selected to continue the BC process.The end product of a BC breeding is a promising line nearly identical to the RP except for carrying the target trait from the donor.Historically,BC breeding had very limited application and was used primarily for improving elite cultivars with respectto disease resistance controlled by single dominant genes.This is because BC plants carrying dominant genes can be directly selected for further backcrossing in each BC generation.When a target trait is controlled by a recessive gene,the breeder must self the heterozygous BCF1plants from each BC generation to identify plants carrying the target recessive alleles for successive backcrossing.For this reason,introgression of recessive genes by phenotypic selection in BC breeding requires twice the time needed for dominant genes.BC breeding was successfully applied[3,4]to improve disease resistance of the elite rice cultivars Pusa Basmati-1 and Kao Dawk Mali 105.The classical BC breeding method has two major advantages.First,the improved cultivar is genetically identical to the RP except for carrying a new trait transferred from a donor,so that extensive yield and adaptation testing is not required before final release.Second,relatively small populations are required for selecting for the target trait because all progeny have a similar genetic background[1-2].As a conservative approach,BC breeding has rarely been used for improving quantitative traits controlled by polygenes.
Advances in DNA markers and genomics since the 1990s have dramatically accelerated progress in genetic analysis and functional genomic studies of genes and QTL affecting a wide range of rice traits.These advances are well described in the Gramene database[5].Several main conclusions can be drawn based on our previous QTL mapping studies using randomly segregating populations[6-11].First,QTL are numerous and widely distributed in the rice genome and their effects are strongly affected by genetic background.Second,the effects of individual QTL on specific phenotypes vary in magnitude,and loci with large effect are rare.Third,most QTL appear to be epistatic,showing varying degrees of interaction with environment.Many large-effect QTL have been cloned by map-based cloning strategies[12-15].These achievements have facilitated the application of marker-assisted BC(MABC)breeding to improving single target traits in crop plants[16,17].MABC breeding offers the same advantages as classical BC breeding,but is much more efficient when foreground selection for target gene(s)and background selection against unwanted donor segments are performed simultaneously[18].However,genetic drag from repulsion linkage between target and undesirable genes in the donor genome can result in failure of genetic improvement[19].It has been suggested[20]that this problem may be solved by two BC generations of genotyping using markers closely(<5 cM on either side)flanking target genes.However,doing so requires genotyping large numbers of BC progeny in each generation in order to identify desired recombinants.As the costs of genotyping have declined in recent years,MABC has been frequently applied to improve many rice traits,including resistances to blast[16,21-23],bacterial blight[24-29],gall midge[30],stripe virus[31]and brown planthopper[32,33];tolerances to abiotic stresses such as submergence[34],salt[35,36],drought[37,38]and phosphorus deficiency[39];and fragrance[40,41].But MABC breeding has limited applications for improving complex traits by introgressing target QTL into elite backgrounds,because doing so requires accurate genetic information about target QTL:magnitude of QTL effects,interactions with other QTL and environment,and linkage relationships with undesirable genes or QTL,which is often unavailable or difficult to obtain.
To unite gene and QTL discovery with practical breeding,Tanksley and Nelson[42]proposed the advanced BC QTL(AB-QTL)analysis method combining QTL analysis with cultivar development.It allows simultaneous discovery and transfer of valuable QTL alleles from unadapted donor lines into elite inbred lines and opens the door to exploiting exotic germplasm for quantitative trait improvement of crop plants.Compared with the QTL detection strategy using conventional mapping populations(F2,BC1,recombinant inbred lines,etc.),AB-QTL analysis offers the following advantages[43-45]:(1)improved accuracy in measuring complex traits in an elite background;(2)improved predictability of QTL behaviors in the target genetic background because of reduced epistasis from the overall lower frequencies of donor alleles in advanced generations;(3)efficient integration of QTL discovery with cultivar improvement;and(4)development of chromosome-segment substitution lines and near-isogenic lines for QTL characterization and cloning.AB-QTL analysis has since been used for genetic dissection of many complex traits of rice,including grain yield[46,47],grain quality[48,49],resistances to biotic stresses[50-52],and abiotic stress tolerance[34,53-57].Like typical genome mapping studies,AB-QTL analysis requires genotyping and phenotyping a large randomly segregating BC population,and is thus difficult to apply in breeding programs involving large numbers of breeding populations.
Recent progress[58,59]in rice functional and population genomic research has made it possible to perform precision breeding,also called genome-and information-based breeding,or breeding by design(BBD)[60].With the explosion of rice genetic and genomic information,the challenge is how to integrate the massive amounts of information with practices of breeding aiming at developing superior cultivars for target environments.In this review,we describe a novel method of breeding by selective introgression(BBSI)for simultaneous genetic dissection and improvement of complex traits,summarizing our efforts over the past 20 years.We explain how BBSI could be implemented in most crop breeding programs to establish the material and information platforms needed for realizing BBD in the near future.
2.Breeding by selective introgression
To overcome the disadvantages and limitations of AB-QTL methods,Li et al.[61,62]proposed a concept and strategy of BBSI for simultaneous improvement and genetic dissection of complex traits.Fig.1 shows a schematic presentation of the strategy of BBSI,which is an improved version of BC breeding but fully integrated with genetic dissection of complex traits by marker-facilitated gene/QTL tracking and allele mining.Depending on the number(single vs.multiple)of target complex traits to be improved,the BBSI strategy has two slightly different versions,each divided into six steps.Step I is to develop randomly segregating BC1F2or BC2F2populations using an elite line as the RP to cross with a set of donors of diverse origins.Starting from the first segregating generation of BC1F1,~25 randomly selected plants from each BC1F1population are selfed or chosen to backcross with the RP to produce~25 BC2F1-family lines.Then,seeds from individual selfed BC1F1or BC2F1lines of each cross are bulk-harvested separately to form a single bulk BC1F2or BC2F2population.In step II,each of the BC1F2or BC2F2populations is separately screened under appropriate conditions with each condition for selecting a single target trait(either abiotic or biotic stress tolerance under stress or yield under non-stress)in direct comparison with the RP(check).Only a very small proportion(2%-5%)of the BC1F2or BC2F2plants that significantly outperform the RP for the target trait are selected.Each BC1F2or BC2F2population will be separately screened for all target traits based on breeding objectives.In step III,all BC1F2or BC2F2plants selected for single target traits of interest are progenytested for all target traits under appropriate stress conditions and for overall performance under normal non-stress conditions.In this step,all single-plant selections are progeny-tested under different conditions for selecting the second target traits and the progeny testing is repeated two to three times depending on the number of additional target traits and the amount of residual genetic variation for the additional target traits in the previously selected lines.By the end of step III,all lines selected from a singleBC population should have been confirmed to carry improved target trait(s)and to be otherwise similar to the RP phenotypically,forming a set of trait-specifci introgression lines(ILs)in the RP background.In step IV,the selected ILs are genotyped with DNA markers or by next-generation sequencing and phenotyped for target traits and overall performance in replicated experiments in target environments for discovering genes and QTL affecting target traits by tracking and characterizing genome-wide responses of the donor segments to selection and genes and QTL affecting non-target traits by linkage mapping or selective introgression(SI).In step V,promising homozygous ILs that carry the target trait(s)and outperform the RP under normal conditions enter preliminary yield trials based on the results of step IV,and the best lines are nominated for national coordinated yield trials(NCYT),and eventually released as new cultivars when they outperform the CK in an NCYT.In step VI,all developed ILs in an elite RP genetic background and their relevant genetic information for target and non-target traits form the material and information platforms for further improving multiple complex traits by designed QTL pyramiding(DQP)or BBD[60].The differences between BBSI and classical or MABC breeding are shown in Table 1.
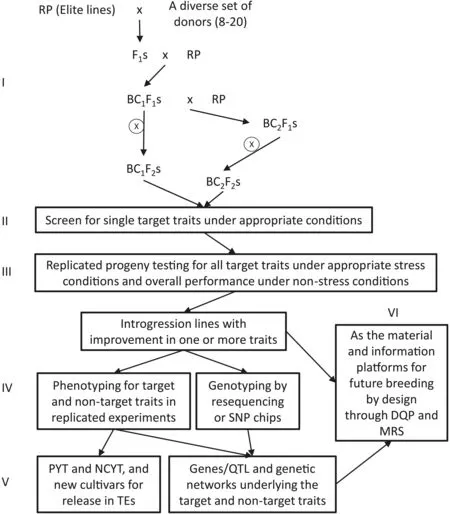
Fig.1.A schematic presentation of the strategy of backcross breeding by selective introgression(BBSI)for simultaneous improvement of single or multiple complex traits.RP,recurrent parent;PYT,preliminary yield trials;NCYT,national coordinated yield trials;TE,target environment;DQP,designed QTL pyramiding;MARS,marker-assisted recurrent selection[60].
2.1.BBSI for improving single complex traits
The strategy of BBSI shown in Fig.1 has been implemented for improving tolerance or resistance of several elite lines to many abiotic and biotic stresses at IRRI and in China[63-70].These include responses to drought,salinity,submergence,phosphorus and zinc deficiency,anaerobic germination,low temperature,and heat,and high grain yield under normal conditions.Table 2 summarizes part of the results from our BBSI efforts,which led us to three important conclusions.First,there are large amounts of useful genetic variation for virtually all complex traits in the primary gene pools of O.sativa,which is largely hidden at the phenotypic level and remained underutilized in past rice improvement.This point could be clearly seen from our BBSI results of cold tolerance(CT)[71]and submergence tolerance(SUT)[72].In the first case,the three xian(indica)donors all had very poor CT but were good donors contributing to the improved cold tolerance of the selected ILs in a geng(japonica)background.In the second case,two donors,TKM9 and Khazar,were highly sensitive to submergence,but were better donors contributing SUT to their BC progenies in three genetic backgrounds than the highly submergence-tolerant donor,FR13A.Apparently,genes/alleles for tolerance to cold and submergence are hidden in these donors.Second,BC breeding plus appropriate phenotypic selection(under appropriate stresses in direct comparison with RPs)is a powerful way to exploit this hidden diversity.Third,donors more distantly related to RPs tend to contribute more transgressive segregation in BC progenies[64,73].

Table 1Similarities and differences between classical BC breeding,MABC breeding,conventional pedigree breeding,and BBSI.
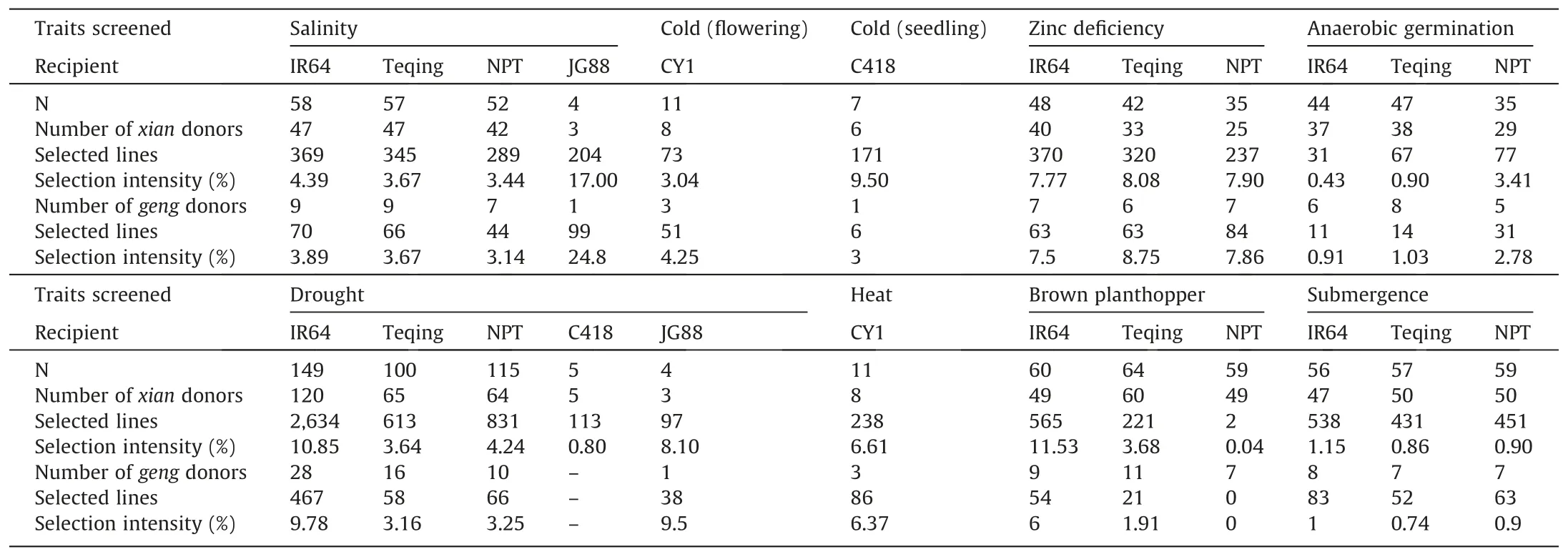
Table 2Summary results of BC breeding efforts for improving tolerance or resistance to eight abiotic and biotic stresses in five elite genetic backgrounds[IR64 and Teqing are xian(indica);NPT,Chaoyou 1(CY1)and Jigeng 88(JG88)are geng(japonica)][73].
2.2.BBSI for improving two or more complex traits
Although BC breeding has been demonstrated to be an effective approach for improving single complex traits,particularly abiotic stress tolerance in rice,it has also been shown useful for improving two or three complex traits by multiple rounds of phenotypicselection on different target traits.Using eight BC1populations derived from a widely adaptable recipient,Huanghuazhan(HHZ),and eight donors of diverse origin with three rounds of phenotypic selection from BC1F2to BC1F4,Ali et al.[74]developed 496 HHZ ILs,most of which showed significantly higher grain yields under one to three abiotic(drought and salt)and/or non-stress conditions(Fig.2).Phenotypic selection schemes implemented using BC1F2bulk populations included a first round of single-plant selections for single target traits(higher grain yield under irrigated conditions,and tolerance to drought and salinity under severe stress conditions)in the BC1F2generation,followed by two rounds of progeny testing of all the selected BC1F3and BC1F4lines across all stress and irrigated conditions.Finally,496 BC1F5ILs were developed from the eight BC1populations,each showing significant improvement for two or more target traits as compared with HHZ.The principle behind BC breeding for improving multiple traits is to exploit the residual genetic variation present in the lines selected from the first and second rounds of screening.At least six of the 496 HHZ ILs were directly released in a short period of six years as new green super rice(GSR)varieties in the Philippines and Pakistan and are now grown in more than 1 million ha in saline,rainfed,or irrigated rice ecosystems[74-76].
The BC breeding component of steps I-III and V of BBSI has been implemented on a massive scale in China and at IRRI to introgress useful traits/genes/alleles into 46 elite lines(commercial cultivars and hybrid parents)from a mini-core collection of 200+accessionsfrom worldwide sources.This activity has resulted in the development and wide adoption of many GSR cultivars that show high and stable yields under reduced input(water,fertilizers,and pesticides)in China and many Asian and African countries[74-76].These efforts have clearly demonstrated the power and advantages of BBSI as a powerful breeding strategy for improving complex traits.Several key results were obtained by our BBSI efforts.First,selecting appropriate RPs is essential for the success of BBSI.RPs should be widely adaptable superior cultivars with high yield potential.Second,the first round of strong single-plant selection in early segregating generations followed immediately by one or two rounds of progeny testing across both stress and non-stress environments in BBSI has two advantages.First,the high selection intensity(typically viability selection)under stress conditions in the first round of selection will instantly remove most progeny lacking the target stress tolerance.Second,the second round of progeny testing of all selected progenies across multiple stress and non-stress conditions will not only validate the effectiveness of the first-round single-plant selections for single target traits,but also allow full exploitation of the residual genetic variation in the selected progeny for other target traits.Thus,two rounds of progeny testing will allow identification of promising ILs carrying desirable genes/alleles for other target traits in multiple testing environments.The presence of residual variation for multiple traits could be seen in Chaoyou 1(CY1)ILs,which showed improved CT and high levels of variation for most yield traits under the normal non-stress conditions[71].More importantly,trait-specific ILs developed from BBSI are unique in that ILs with the same elite genetic backgrounds were phenotypically similar to their RPs but each carried one or a few traits from a known donor,and together they harbored a high proportion of loci/alleles for improved target traits from diverse donors.Thus,these ILs are useful materials for efficient genes/QTL discovery and for mining desirable alleles affecting complex traits for future BBD[60,62].
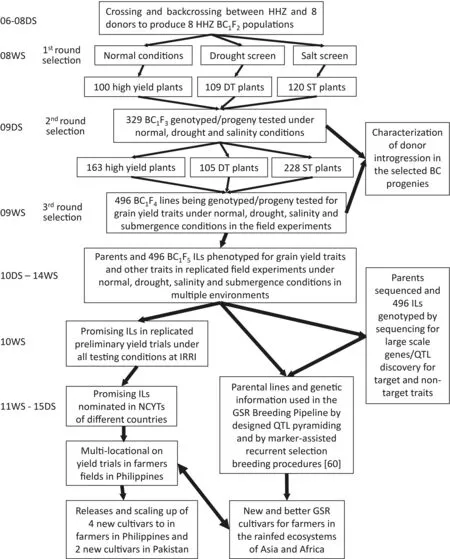
Fig.2.A scheme of breeding by selective introgression(BBSI)to develop GSR cultivars with improved tolerance to multiple abiotic stresses(drought and salt)using eight Huanghuazhan(HHZ)BC1 populations[74].BC,backcross;DT,drought tolerance;ST,salt tolerance;ILs,introgression lines;GSR,green super rice;NCYT,national coordinated yield trials.
2.3.Efficient identification of genes/QTL and QTL networks affecting target and non-target traits using ILs and DNA markers
Step IV of BBSI is to dissect genes/QTL and genetic networks underlying target and non-target traits using the selected ILs and DNA markers(Fig.1),another major advantage distinguishing BBSI from conventional breeding procedures that do not generate genetic information for any traits.The purpose of this step is to establish an information platform for future development of superior cultivars by DQP and BBD using ILs.
The theoretical basis for dissecting genes/QTL and genetic networks underlying target traits in the ILs is based on population genetics theory[77,78].Because a set of ILs is selected for improved(relative to the RP)target traits from a BC population,these traits must be associated with donor alleles at specific loci affecting the traits.Thus,the donor alleles at loci associated with the target traits in the selected ILs will be present at higher frequency than those theoretically expected in unselected BC populations.The higher the frequency of the donor allele at a specific locus(QTL)in the ILs,the greater is the effect of the donor allele on the target trait.Statistically,detecting maineffect genes or QTL affecting target traits in ILs selected from a specific BC population using DNA markers requires performing genome-wide tests to identify loci at which donor allele frequencies exceed their theoretical expectations in unselected populations,using eitherχ2tests[61]or Wald tests of segregation distortion(WSD)[79].Withχ2tests,the expectations should be obtained from the unselected BC population from the same cross as the selected ILs and not the Mendelian expectation,given that introgression frequencies of different donors in specific RP genetic backgrounds in different BC generations may vary considerably and deviate significantly from Mendelian expectations.Statistically,the WSD is superior to theχ2test because it employs a joint analysis of ILs selected from several populations derived from a common RP and multiple donors using a general linear model[79].
Selection may cause strong nonrandom associations among donor alleles at unlinked main-effect loci involved in epistasis affecting target traits[80,81].Thus,ILs developed by strong selection for target traits in a BC population are useful materials for detecting epistatic QTL affecting target complex traits[82].Classically,nonrandom associations among unlinked main-effect loci(detected by the segregation-distortion tests described above)can be quantified by gametic or zygotic linkage-disequilibrium(LD)statistics.Considering two unlinked loci A and B,the gametic LD statistic,D^AB=pAB-pApB,is normally calculated using the genotypic data of ILs from each BC population,where p~A,p~B,and p~ABare the relative frequencies of two unlinked loci,A and B,and their digenic genotype AB,respectively[83].For the zygotic LD statistic,Dijkl=pijkl-pijpkl=nijkl/n-nijnkl/n2,where nijk,nij,and nklare the numbers of genotypes ijkl(i,j=A,a;k,l=B,b),ij(i,j=A,a),and kl(k,l=B,b)in a sample of size n[84].However,when the number of segregating loci involved in epistasis affecting a target trait is >3, a multi-independence probability test,P(AG)=(pi)rm· (1-pi)r(n-m),can be used to detect nonrandom associations among a group of unlinked epistatic loci(an association group,or AG)resulting from strong selection,where P(AG)is the probability of a group of r unlinked loci that are perfectly associated with one another in ILs selected for a target trait from a BC population,piis the expected frequency of the donor introgression at the i th locus in the ILs from the BC population(i=1,2,3...r),n is the number of ILs selected for the target trait from the BC population,m is the number of ILs that carry a co-introgression of the donor alleles at the r unlinked loci,and(n-m)is the number of ILs carrying no introgression at the r unlinked loci in the AG.Theoretically,an AG is defined as a group of loci acting epistatically in the same signaling pathway affecting the target trait segregating in the BC population[82].
2.4.Dissecting genetic loci and networks underlying target traits using ILs
In the past few years,BBSI has been successfully used to dissect genetic networks underlying rice tolerance to several abiotic stresses using ILs and methods described above.
2.4.1.QTL networks for drought tolerance(DT)
Using 72 ILs selected for improved yield under drought stress from four large BC2F2populations derived from crosses between an elite geng variety,Jigeng 88(JG88)(RP),and four donors,Cui et al.[85]reported putative genetic networks(multi-locus structures)consisting of 30 loci(main-effect QTL)in 29 functional genetic units(FGUs),each defined as either a single main-effect locus or an AG,underlying DT byχ2and multi-locus probability tests.Fig.3A shows a genetic network consisting of eight FGUs in two independent branches plus a single locus(RM541 in bin 6.5)for DT detected in 21 DT ILs selected from a JG88/MR77 BC2population.Branch A-1 has four positively correlated FGUs with RM449 in bin 1.1 as a putative regulator and three downstream FGUs,agDtII-1(RM336 in bin 7.6 and RM331 in bin 8.3),RM286(bin 11.1),and RM470(bin 4.6),while branch A-2 has three FGUs positively associated with RM406 in bin 2.8 as a putative regulator and RM276(bin 6.3),and RM311(bin 10.3)in the downstream of the branch[85].
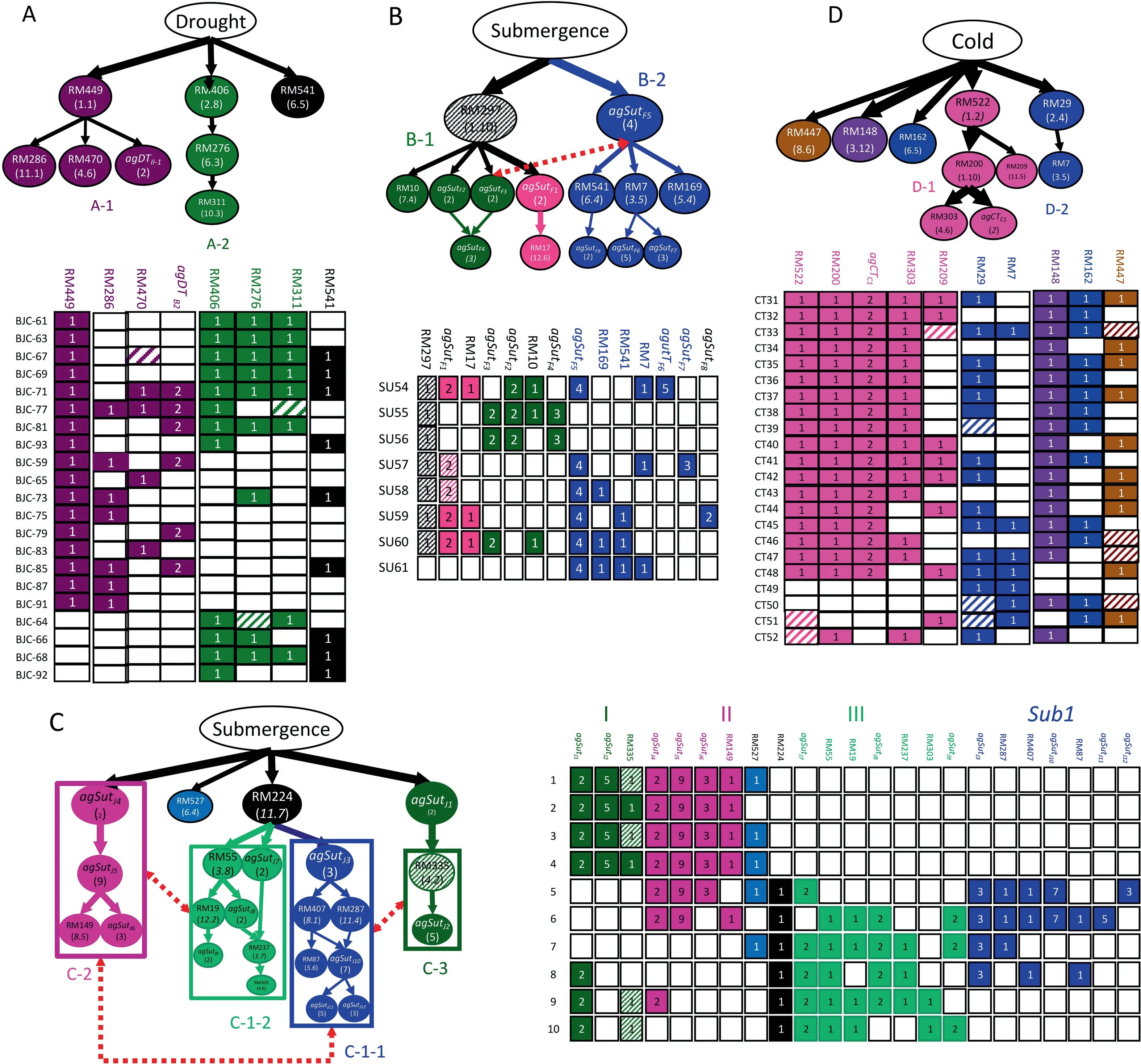
Fig.3.Putative genetic networks(multi-locus structures)underlying tolerances to drought,submergence,and cold detected in introgression lines(ILs).(A)A genetic network consisting of eight functional genetic units(FGUs)(nine loci)for DT detected in 21 DT ILs from the JG88×MR77 BC2 population[85].(B)A genetic network consisting of 14 FGUs(29 loci)detected in eight SUT ILs from the Teqing×FR13A BC2 population[72](with permission from John Wiley and Sons).(C)A genetic network consisting of 23 FGUs(56 loci)in three major groups detected in 10 SUT ILs from the NPT×TKM9 BC3 population[72].(D)A genetic network consisting of 10 FGUs in three major groups detected in 22 CT ILs from the CY1×Fengaizhan BC2 population[86].In the graphical genotypes corresponding to each network,the unfilled,fully colored,and hatched cells represent recurrent-parent homozygote,donor homozygote,and heterozygote genotypes.The number in the cells of each FGU is the number of loci included in the FGU.Solid arrow lines connecting two FGUs in each branch of a network represent putative functional relationships with those of high introgression as putative upstream regulators and those of low introgression in the downstream of a branch.The thickness of an arrowed line is proportional to the introgression frequency of the downstream FGU.
2.4.2.QTL networks for SUT
Using 162 SUT ILs selected from 12 BC populations derived from crosses between three RPs,IR64,Teqing,and NPT and three donors,TKM9,FR13A,and Khazar,Wang et al.[72]reported the identification of 12 genetic networks or multi-locus structures each containing many FGUs affecting SUT in the ILs selected from one of the 12 BC populations.Together,the 12 networks included 295 maineffect loci in 167 FGUs across 68 bins of the rice genome.Fig.3B shows a genetic network consisting of 14 FGUs(29 main-effect loci)in two major branches detected in eight BC2SUT ILs selected from the Teqing×FR13A BC2population.Branch B-1 contains seven FGUs positively associated with RM297(bin 1.10)as the putative regulator and six downstream FGUs,including agSutF1(two loci),agSutF2(two loci),agSutF3(two loci),agSutF4(three loci),RM10(bin 7.4)and RM17(bin 12.6).Branch B-2 contains seven FGUs positively associated with agSutF5(four loci)as the putative regulator plus six downstream FGUs,RM541(bin 6.4),RM7(bin 3.5),RM169(bin 5.4),agSutF6(five loci),agSutF7(three loci),and agSutF8(two loci).Fig.3C shows a genetic network consisting of 23 FGUs(56 main-effect loci)in three major groups detected in 10 SUT BC3ILs from the NPT×TKM9 BC3population.In branch C-1,the highest introgression frequency(IF),RM224(bin 11.7)was placed at the top as the putative regulator highly associated with two major sub-branches.agSutJ3(bins 5.4,6.2,and 8.6)was inferred to be the putative regulator of sub-branch C-1-1 based on its higher IF,and highly associated with six downstream FGUs(bins 8.1,11.4,5.6,agSutJ10,agSutJ11,and agSutJ12)of lower IF.In sub-branch C-1-2,bin 3.8 and agSutJ7(bins 6.2 and 11.5)had the highest IF and were placed at the top as putative regulators,being highly associated with four downstream FGUs(bins 12.2,4.6,1.7,agSutJ8and agSutJ9)of lower IF.In branch C-2,agSutJ4(bins 4.7 and 10.7)had high IF and was assigned as the putative regulator,associated strongly with agSutJ5(bins 1.3,2.5,2.11,6.6,7.5,9.3,9.6,10.3,and 12.6),agSutJ6(bins 4.2,6.4,and 8.3),and RM149(bin 8.5)of lower IF in the downstream of branch C-2.In branch C-3,agSutJ1(bins 5.2 and 10.2)had high IF and was assigned as a putative regulator associated strongly with bin 4.2 and agSutJ2(bins 1.5,3.9,5.5,7.6,and 12.4)of lower IF in the downstream of branch C-3.Strong negative associations were detected between the downstream FGUs of branch C-1-1 and those of branch C-3,and between branch C-1-2 loci and the downstream loci of branch C-2[72].Most importantly,it is clear that each of the selected SUT ILs has a graphical genotype containing multiple identified FGUs.
2.4.3.QTL networks for CT
Using the same strategy with 30 CT BC2ILs developed from two rounds of selection and progeny testing from four large BC2F2populations from crosses between a superior geng restorer line,C418(RP)and four cold-sensitive xian lines(donors),Zhang et al.[69]characterized genome-wide responses to strong phenotypic selection for CT at the vegetative stage in rice and reported four genetic networks consisting of significantly over-introgressed donor alleles at 56 main-effect loci in 28 FGUs across the rice genome.Using 84 CT ILs from five BC2populations of CY1(RP)and five diverse but cold-sensitive donors developed after two rounds of strong selection for CT at the reproductive stage,Liang et al.[86]characterized QTL networks consisting of 46 FGUs(50 main-effect loci)underlying rice CT at the reproductive stage.Fig.3D shows a genetic network consisting of 10 FGUs(one AG and nine loci)with two major branches plus three independent loci detected in 22 CT ILs from the CY1×Fengaizhan BC2population.Branch D-1 was the most important branch,consisting of five FGUs(one AG and four loci)positively associated with RM522(bin 1.2,IF=0.91)plus four downstream FGUs,RM200 in bin 1.10,agCTC1(bins 2.8 and 9.7),RM303(bin 4.6)and RM209(bin11.5).
2.4.4.The power and efficiency of BBSI for genetic dissection of abiotic stress tolerance in rice
Table 3 summarizes recent efforts of BBSI in genetic dissection of rice tolerance to abiotic stresses(drought,submergence,and cold)using 351 ILs developed from 25 BC populations.The BBSI strategy offers four major advantages in genetic dissection of abiotic stress tolerance over conventional QTL-mapping approaches using large randomly segregating populations.First,BBSI is much more powerful and efficient with respect to the number of detected loci,the possibility of allele mining,and the power of detection of high-order epistasis.This superiority is evidenced by the identification of 110.2 QTL(45.8 single loci and 21.5 AGs)affecting one abiotic stress tolerance,or 17.7 detected QTL(7.3 main-effect QTL and 3.4 AGs)/trait/population with a mean population size of 14.4 ILs/population,by the identification of many AGs consisting of 3-10 unlinked but perfectly associated loci,and by the possible presence of multiple alleles from 3 to 5 different donors at detected loci(Table 3).Second,the genetic(QTL)networks underlying DT,SUT,and CT,and the graphical genotypes of the identified QTL for target traits in corresponding DT,SUT,and CT ILs obtained by BBSI(Fig.3),provide information about the genetic relationships between or among detected loci that are consistent with our knowledge that plant abiotic stress tolerances involve many signaling pathways and complex gene networks.For example,the networks(Fig.3B,C)underlying SUT with two or four groups of loci each consisting of multiple unlinked but highly associated FGUs showing a clear hierarchy of magnitudes (introgression frequencies)most likely represent loci involved in a signaling pathway affecting SUT in rice.The indirect but convincing evidence for this interpretation came from the observation that three of the four loci of agSutF5,the inferred regulator of branch B-2 detected in the BC2population Teqing×FR13A and the three loci of agSutJ3,the inferred regulator of branch C-1-1(both in blue color)detected in BC3population NPT×TKM9 share the same genomic locations near bins 6.2 and 8.6[72].The wellknown Sub1A gene cloned from FR13A is located in bin 9.6 and is an ethylene-responsive factor controlling rice SUT[87].In fact,the three-or four-locus AG containing Sub1A was completely or partially detected in SUT ILs from seven of the 12 BC2populations we investigated[72].Thus,loci of branch B-1 and C-1-1 can be confidently inferred to be involved in the ethylene-regulated pathway(s)controlling rice SUT.The results in Fig.3B and 3C also suggest that Sub1A alone is necessary but insufficient for a high level of SUT in rice.Third,large numbers of loci involved in the genetic networks suggest that genetic complementarity and repulsion-phase status of the functional alleles in the parents explains the observed hidden diversity and transgressive segregation for abiotic stress tolerance of rice(Table 2).Most importantly,the graphical genotypes of the ILs for the identified loci affecting target traits provide direct information for BBD revealing how the improvement in the target trait of each of the ILs was achieved via combinations of the detected QTL.
2.5.Identification of QTL affecting non-target traits using ILs
In addition to affording high power and efficiency for genetic dissection of target traits,the ILs developed by BBSI can be used for detecting genes or QTL affecting non-target traits.Because of strong phenotypic selection,the number of ILs selected for a single target trait from a single BC population is usually too small to detect QTL affecting non-target traits.However,each BC population of a BBSI program will normally be selected for multiple target traits and the number of BC populations(donors)used for improving any given RP in a BBSI program is moderately large,resulting in hundreds of ILs with improvement of multiple target traits in an elite genetic background,particularly when BBSI for multiple target traits is implemented.Thus,ILs developed by BBSI can be used for identifying QTL affecting non-target traits using a random model,y=Xβ+Zkγk+ξ+ε,where Zkis an indicator of genotyperepresented by a n×m matrix for locus k,n is the population size and m is the number of founders,γkis a m×1 vector for the m founders’allelic effects,ξis the polygenic effect andεis the residual error[88].A Wald test statistic with a chi-square distribution with m-1 degrees of freedom can be calculated for each marker along with a corresponding P-value.

Table 3Summary of power and efficiency of detecting QTL affecting abiotic stress tolerance in rice by BBSI.
2.5.1.QTL for bacterial blight resistance(BBR)and CT
Using the abovementioned 496 HHZ ILs selected for multiple abiotic stress tolerances from 8 BC1populations[74],Xu[89]identified 65 QTL associated with resistance to 14 Philippine strains of Xanthomonas oryzae pv.oryzae(Xoo),which causes rice bacterial leaf blight.The identified resistance QTL explained 5.3-68.4%of the total phenotypic variance in lesion lengths in the combined HHZ IL population,including one major resistance QTL conferring high-level resistance(lesion lengths<0.5 cm)to all 14 Xoo strains.Using an F2population derived from a cross between FF329(an IL with high resistance)and HHZ,the complete resistance of FF329 was confirmed to be controlled by a single dominant gene,designated Xa39[90].Using the same 496 HHZ ILs and replicated phenotyping under natural low temperature(17-20°C)stress during the reproductive stage and 41,754 high-quality single nucleotide polymorphisms derived from genotyping by resequencing,Zhu et al.[91]identified six QTL on chromosomes 3,4,and 12 that affected CT at the booting to milky stages of rice.One QTL,qCT-3-2,which conferred stable CT across years and locations,was finemapped to a genomic region of 192.9 kb.
2.5.2.QTL for yield traits
Three sets of 125 BC2F4ILs from single-plant selections for high yields from BC2F2populations derived from crosses between SH527(RP,an elite restorer)and three donors(ZDZ057,Fuhui 838,and Teqing)were used for identification of QTL affecting yield traits in rice[92].The 125 ILs were evaluated for eight yield traits in replicated experiments across the short-day environment in Hainan and the long-day environment in Anhui.Because the mean trait values of the ILs were not different from those of the RP and all measured traits showed typical normal distributions with transgressive segregation in both directions,both BBSI and classical QTL mapping were applied,resulting in the identification of 94 QTL affecting the eight yield traits,or~2 QTL per trait-/population/environment combination with a mean population size of 41.7.Of the 94 QTL detected with the same statistical threshold of P<0.001,88 were detected by the BBSI method,while only six were identified by the classical linkage mapping approach[92].
Our results suggest at least three advantages of using ILs to identify QTL affecting non-target traits:(1)more accurate phenotyping in replicated experiments across multiple environments because of the relatively small population sizes of ILs and similar genetic backgrounds,(2)cost saving in both genotyping and phenotyping because of the relatively small population sizes of ILs,and(3)increased power resulting from(1)and combined population analyses[79].Statistically,the false-positive rate is expected to be very low for QTL affecting non-target traits identified with the same threshold,based on data from small selected populations in replicated experiments[93].
3.Simultaneous improvement of multiple traits by DQP
The final step of BBSI in the original design of Fig.1 is to use the trait-specifci ILs and their phenotypic and genetic information about target and non-target traits obtained from steps III and IV to develop superior cultivars by DQP.Here,DQP is a vital strategy for realizing BBD in the future.In fact,DQP has been implemented,though not completely as originally designed,for improving high yield and multiple abiotic stress tolerances in rice[88,94,95].Based on phenotypic performances of ILs and favorable alleles at QTL detected for target and non-target traits,it is easy to pyramid favorable alleles for multiple target traits from different donors in the same elite backgrounds by MAS and/or phenotypic selection.Feng et al.[88]selected two HHZ ILs,M79 and M387.M79 carries two major QTL,qDT3.9 and qGY1,conferring improved tolerance to drought and low-nitrogen treatment(LNT)from the donor,OM1723(a xian cultivar from Vietnam).Similarly,M387 carries two different major QTL,qDT6.3 and qSF8 conferring improved DT and LNT from Teqing,a donor from South China.MAS using four pairs of Kompetitive Allele-Specific PCR markers,combined with phenotypic selection on an F2population from cross M79×M387,resulted in the rapid identification of four promising lines,each pyramiding favorable alleles at three of the four target QTL.The four lines showed stronger tolerance to drought and/or low nitrogen[94].Using the same DQP strategy by crossing two HHZ ILs with good yield potential and improved tolerances to drought and submergence,Pang et al.[95]successfully developed six promising lines with improved salt tolerance and yield potential relative to the parental ILs.
4.Summary,challenges,and perspectives
Today,the greatest challenge facing plant breeders is the major shift in breeding objectives from emphasizing yield improvement to achieving sustainable yield improvement and stability with efficient use of resources.Diverse requirements for grain quality add another dimension of difficulty to meet future food demands under increasingly frequent and extreme environmental disturbances resulting from global climate change[60,76].To meet this challenge,future breeding technology using BBD must be built upon the massive information gained from progress in crop functional and population genomics,and BBSI is a powerful strategy designed to meet the challenge by building the material and information platforms required for BBD.Attentions should be given to four concerns to achieve the best results of BBSI.
First,the primary purpose of steps I-III of BBSI is to develop hundreds and thousands of ILs in elite genetic backgrounds as the material platform for future BBD.Thus,in addition to selection as RPs of the‘‘best”lines with wide adaptability to target environments,the use of 200-300 mini-core accessions of the world crop germplasm collection as donors is key to ensure that each set of ILs in an elite background contains the‘‘best’”alleles at large numbers of loci affecting any target traits of interest introgressed from the donor gene pool to be identified in the target environments.This strategy would greatly increase the functional(phenotypic)predictability of donor alleles in the further BBD process.Second,effective phenotypic selection for target traits is necessary.Empirically,the size of the initial segregating BC population for the first round of single-plant selection should be 400-600 so that it is relatively easy to apply a severe(nearly lethal to RPs)stress at critical developmental stage(s)to identify superior BC progeny by direct comparison with RPs.This will increase the accuracy of selection and reduce the number of selected plants to a manageable size in the early BC generations.However,it is not clear whether and how phenotypic selection for target traits can be effectively implemented in off-season winter nurseries,a common practice used to speed up breeding processes.How differences between the offseason and target normal-season environments will affect the selection efficiency and expression of genes or QTL affecting different target traits is a question for future investigation.Third,in addition to its much improved power and efficiency,the greatest advantage of BBSI over classical gene and QTL mapping approaches is that all genetic information(numbers,genomic locations,andgenetic relationships of genes and alleles)for both target and nontarget traits is directly applicable to real breeding populations because all of these are obtained in the target genetic backgrounds and environments.It should be pointed out that the genetic networks constructed based on strong nonrandom associations between or among unlinked loci in selected ILs in Fig.3 should be interpreted cautiously.Theoretically,unlinked but strongly associated loci in an AG detected in ILs selected for a target trait should be those with mutual functional dependence and thus epistatic to one another[80-82].However,nonrandom associations among unlinked loci could also result from sampling error,given the small numbers of selected ILs,a particular danger for loci with low donor introgression in ILs.There are two ways to validate the inference that a detected AG or a branch of associated loci is a genetic network.A straightforward way is to determine whether the nonrandom associations among unlinked loci persist in progeny of the DQP F2population selected for the same target traits.In fact,the validation of genetic networks affecting target traits is part of the purpose of DQP or BBD experiments.The second approach is to clone most,if not all,loci in a genetic network and to determine their functional relationships at the molecular level.This approach has been partially demonstrated in several cases by cloning the regulatory genes acting upstream of genetic networks affecting complex traits[14,15,87,96].However,molecular cloning of downstream QTL in the genetic networks would be technically difficult and can probably be achieved by multidisciplinary approaches.In contrast to the successful implementation of the BC breeding component of BBSI in China and at IRRI[63-69],it has been a major challenge to fully implement step IV of BBSI for large scale gene or QTL discovery and allele mining for both target and non-target traits in the massive BC breeding efforts of rice in China,largely because of the absence of the capacity required to do so in most small breeding programs in the past.Fortunately,this component of BBSI is expected to be greatly facilitated and accelerated by rapid cost reduction in genotyping by sequencing and by the public availability of various types of genomic diversity in rice populations[59,97,98].
Finally,DQP in step VI of BBSI,as briefly described above,is a prototype of BBD for efficiently developing superior rice cultivars by improving multiple complex traits.The idea is simply to pyramid,via either MAS with genotyping by sequencing,genomic selection,or phenotypic selection,‘‘best”alleles at many target loci from two or more donors in the same elite genetic backgrounds based on accurate genetic information about genes and alleles affecting different target traits obtained in step IV[63,94,95,99].A more comprehensive strategy of MARS built upon ILs and the relevant information platform facilitated by a dominant-gene controlled male sterility system has been proposed for realizing future BBD in self-pollinated crop species[60]and has been tried in rice[99],but remains to be fully demonstrated.
With respect to future perspectives,two major advances have signaled the readiness of rice breeding in China to enter the era of genome-based BBD.First,thousands of trait-specific ILs in 30+elite genetic backgrounds that contain huge amounts of useful genetic diversity for various target traits from a mini-core germplasm collection of rice have been developed in various Chinese institutions(our unpublished results).Second,the expected rapid progress in global functional and population genomic research in major crops will eventually reveal all functional variation in the rice gene pools,which has been or is being integrated into various databases.Furthermore,the strategy of using selected ILs and marker-assisted characterization of genome-wide responses to selection can be easily extended for efficient gene and QTL discovery and allele mining of complex traits using advanced(F3-F8)breeding lines developed by the conventional pedigree breeding approach,though crosses should be made between a backbone parent and a set of different elite parents to avoid possible severe linkage drag from distantly related landraces.Finally,the proposed strategy of BBSI can be readily adopted by relatively small breeding programs in major crops aiming at a single target environment,based on our experience in rice.In fact,the full implementation of a BBSI program in Fig.1 can be easily adopted by a small breeding program aiming at developing superior cultivars with improved tolerance or resistance to four abiotic and biotic stresses plus desirable grain quality and yield potential for a specific target environment.It will take 6-8 years to develop,from a set of minicore germplasm accessions,IL population(s)in elite RP background(s)that contain large numbers of desirable genes and alleles affecting a wide range of target traits.The scale of activities and costs for population development,target-trait screening,phenotyping,and genotyping(in house or by outsourcing)the selected ILs should be within the capacity and budgets of most seed companies and institutions in China,given the increasing trend of phenotyping cost and declining trend of genotyping cost.The major limitations to implementing a successful BBSI will then remain the needed software pipeline for data analyses in steps IV and VI of BBSI and well-trained technicians.
CRediT authorship contribution statement
Zhikang LiandJianlong Xuconceived and designed this manuscript;Fan ZhangandYingyao Shiwrote the manuscript;Jauhar Alirevised the manuscript.All authors read and approved the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was funded by the National Key Research&Development Program of China(2017YFD0100100),Key-Area Research&Development Program of Guangdong Province(2020B020219004),Shenzhen Basic Research Special Project(2020231601),and Agricultural Science and Technology Innovation Program and the Cooperation and Innovation Mission(CAAS-2021-01).We thank Prof.James C Nelson,Kansas State University,for editing the English text of this manuscript.

- The Crop Journal的其它文章
- Breeding by design for future rice:Genes and genome technologies
- Innovation and development of the third-generation hybrid rice technology
- Understanding the genetic basis of rice heterosis:Advances and prospects
- CRISPR/Cas systems:The link between functional genes and genetic improvement
- Genomic selection:A breakthrough technology in rice breeding
- Target chromosome-segment substitution:A way to breeding by design in rice