
胺脱氢酶在手性胺生物合成中的研究进展
2021-06-17 05:47郑晨妮陈蕊蕊王志国陈振明
杭州师范大学学报(自然科学版) 2021年3期
郑晨妮,吴 刚,贺 瑾,陈蕊蕊,王志国,陈振明
(1. 杭州师范大学生命与环境科学学院,浙江 杭州 311121; 2. 浙江海森药业股份有限公司,浙江 东阳 322100;3. 杭州师范大学医学部,浙江 杭州 311121)
0 前言
手性胺是众多小分子药物、农药和精细化学品的重要前体,常用于合成各种药物活性物质和化学品.据估计,在最常用的200种小分子处方药物中,超过三分之一含有手性胺结构[1],如洛匹那韦-HIV蛋白酶抑制剂、用于治疗阿尔茨海默病的卡巴拉汀以及镇痛剂可待因等[2-3].目前,手性胺的制备主要应用化学合成法和酶催化合成法.化学合成法需要多种危险、有毒的试剂和重金属催化剂,并且反应步骤多、反应条件苛刻,导致手性胺产率较低并伴随严重的环境污染[4],同时由于产物后处理困难,生产的胺手性物质纯度很难达到工业生产要求.如在N-乙炔酰胺的合成路线中,需在-10 ℃以下进行部分反应,然后在70 ℃进行肼解[2].近期研究发现,借助酶催化反应,手性胺的合成可以在温和的条件下进行,具有较高的对映选择性、转化率和工业规模的时空产率.同时酶也是一种低危害的催化剂,能够从自然可再生资源中获得,并可以通过固定化形式进行回收利用[5].由于酶具有活性高、特异性强、立体选择性高、反应条件温和等优势,生物酶法逐渐取代化学合成方法成为手性胺制备中最有前景的合成方法之一.
近年来,转氨酶[6]、裂解酶[7]、亚胺还原酶[8]、胺脱氢酶等已经成功用于手性胺的合成.其中转氨酶能够将氨基供体上的氨基转移到羰基受体上,从而形成胺活性物质.野生型转氨酶的结构特异性和立体选择性均较低[9].在转氨酶的反应过程中涉及氨供体的参与,由于不同构型的酶需要不同构型的氨供体,导致供体范围有限.因此,氨供体、氨供体与酶的亲和力以及辅酶成为应用转氨酶制备手性胺的限制性因素.此外,转氨酶反应中还需考虑副产物的去除,进一步限制了转氨酶的应用.裂解酶在手性胺的制备中也有较多应用,如L-天冬氨酸、丙氨酸的合成.但是裂解酶在某些反应如L-苯丙氨酸的合成[7]中不能介导C—N键的形成.另外,裂解酶的最适pH与级联酶差异过大,不能用于“一锅”反应,严重限制了其广泛应用.亚胺还原酶活性低,催化机理不明确,不能进行有效的半理性设计,且底物范围小[10-11],难以应用于工业生产.相比之下,胺脱氢酶结构特异性高,具有高立体选择性、高原子转化率等优势,有望成为工业化生产中绿色合成手性胺的关键酶.本文重点概述胺脱氢酶在手性胺生物合成中的辅酶循环方式,以及不同类型的胺脱氢酶在手性胺合成中的研究进展,同时展望了其在工业生产领域的应用前景.
1 胺脱氢酶合成手性胺
胺脱氢酶能够以低廉的材料如氨气作为氨供体将酮还原胺化,并且副产物只生成水[12].该催化反应具有立体选择性好、原子经济性高等优点,因此被广泛用于手性胺的合成.然而,此酶催化反应也存在诸多不足:首先,所有的胺脱氢酶都是单向反应,当辅酶消耗殆尽时,反应也会随之停止.而在反应中使用辅酶因子,增加了反应成本,同时反应平衡也是一大问题.其次,胺脱氢酶对于很多底物的胺化活性都比较低,严重限制其工业化应用.本文拟从胺脱氢酶反应平衡,以及不同类型的胺脱氢酶在手性胺合成中的应用两方面进行论述.
1.1 胺脱氢酶反应平衡的构建
1.1.1 胺脱氢酶-丙氨酸脱氢酶级联
在手性胺的合成过程中,胺脱氢酶可以将外消旋底物氧化脱氨形成单一构型的手性胺.此反应需要辅酶NAD+的参与才能进行,而其昂贵的价格无疑会增加反应成本.Patil等[1]通过胺脱氢酶与丙氨酸脱氢酶级联实现了辅酶循环和手性胺的合成.如图1所示,在(R)-型胺脱氢酶作用下,外消旋底物被氧化脱氨形成对应的酮,留下(S)-型手性胺.同时,在丙氨酸脱氢酶的作用下使NAD+得以再生.图2给出了双酶级联体系中具有反应活性的消旋胺.

图1 双酶级联将外消旋胺氧化脱氨生成手性胺

图2 AmDH-AlaDH双酶级联体系中有活性的消旋胺
1.1.2 胺脱氢酶-甲酸脱氢酶级联
胺脱氢酶不止能拆分外消旋胺获得单一构型的手性胺,还能利用氨气及其他物质作为氨供体形成二级和三级胺,其机理是烟酰胺辅酶的氢负离子先转移到氨供体上形成亚氨基中间体,再在酶的作用下将氨转移给氨受体形成手性胺[13].如图3所示,胺脱氢酶能够把不同氨供体(B—H)中的氨基基团转移至氨受体(1—6)上,将其转化为次级手性胺化合物.可以通过向反应体系中加入来自Candidaboidinii的甲酸脱氢酶(Cb-FDH)带动辅酶循环,持续提供还原力.以甲酸或甲酸铵作为辅酶循环原料,反应副产物只有易于去除的CO2,故该手性胺合成工艺成本低廉且更加环保.

图3 AmDH/Cb-FDH级联催化羰基化合物还原胺化
1.1.3 胺脱氢酶-乙醇脱氢酶级联
越来越多的醇类化合物被认为是金属催化胺化法合成胺类化合物合适的起始底物,但金属催化胺化反应立体选择性较差、原子效率较低,且需要使用重金属催化剂,污染性高,严重限制了其应用[14].而胺脱氢酶介导的多酶级联反应可以将乙醇转变为手性胺,反应主要分两步:第一步,乙醇脱氢酶将乙醇转变为相应的酮类物质;第二步,在胺脱氢酶作用下,将氨供体上的氨转移到酮上,从而生成手性胺.
Liu等[15]将依赖NAD(P)+的乙醇脱氢酶与参加辅酶循环的酶共表达,在细胞内循环辅酶并将醇转化为酮类物质,随后在细胞外再进行一次辅酶循环,利用胺脱氢酶将酮转变成手性胺(图4).这种细胞内与无细胞系统偶联的合成方式,将胞内和胞外辅因子的酶循环相互分离,实现了外消旋醇向手性胺转化的级联反应.此级联反应中的限制因素是参与辅酶循环的酶,通过在胞内共表达(S)-和(R)-选择性的ADH和对NAD+具有高转化率的NOX酶,胺脱氢酶和乙醇脱氢酶级联能够将化合物(R)-和(S)-1a-b转化为手性胺(R)-3a和(R)-3b,其反应的ee值为99%,转化率分别为70%和48%,反应过程中NADH循环次数分别是350次和240次.当共表达(S)-和(R)-型ADH、NOX,同时将胺脱氢酶与葡萄糖脱氢酶固定化,级联反应过程中NADH的循环次数可达到1 400次,而生成产物(R)-3a的ee值为99%且其转化率提升到71%~84%,反应路线见图4.

图4 分隔式多酶级联反应合成手性胺
1.1.4 级联反应小结
由上可知,胺脱氢酶反应是不可逆的,且需要辅酶NAD+的参与,提高了制备手性胺的反应成本.这就要求将辅酶循环与胺化反应级联,实现“一锅”反应,加大原子的利用率同时避免副产物的生成,从而更加高效与环保[16].
上述级联反应中,丙氨酸脱氢酶、甲酸脱氢酶、葡萄糖脱氢酶和乙醇脱氢酶利用廉价的底物(甲酸铵、葡萄糖、丙氨酸等)进行辅酶循环,同时循环过程中产生的副产物易于除去,使得反应向正向移动从而持续生成手性胺.尽管通过酶级联反应制备手性胺存在诸多优势,目前仍有一系列问题亟待解决,如:1)反应需在一定的pH和温度下进行,但是不同酶的最适pH和最适温度有所差异.可以考虑从载体构建入手,构建融合蛋白并建立其最佳的反应条件.另外,甲酸的使用为反应体系的优化提供了良好的选择,这是因为很多酶对甲酸有很高的耐受性,并且甲酸价格低、产量大,适于大规模工业生产应用.2)外消旋的胺底物及某些酮类化合物会对酶产生抑制作用.针对此问题,可以将酶固定化,然后分层次进行反应,使整个体系运动起来,避免底物或产物因浓度过高而抑制酶的活性.3)辅酶循环的效率问题是最大的限制因素.可以考虑优化循环辅酶酶稳定的反应条件,保证循环辅酶酶的活性,然后在合适反应条件下进行相关反应.另外,级联的酶的活力差异会造成辅酶循环“间断”,导致整个反应速度下降,此时可以对催化辅酶循环的酶进行突变,提高对辅酶的活性,从而提高辅酶的循环效率,或是将两种酶固定化,然后在细胞外构建一个新的环路反应,也可将反应在细胞内和细胞外同时进行,加速辅酶的循环,从而提高手性胺的转化率.
1.2 基于氨基酸脱氢酶的胺脱氢酶
越来越多的酶被用于合成手性胺,这种绿色、新颖的合成方式正逐步取代利用铑作为催化剂的化学合成法.酶法需要用到氨源作氨供体,还涉及反应平衡问题.胺脱氢酶是合成手性胺的最有潜力的生物催化剂之一,能够单向将酮类化合物转化成手性胺,不涉及动力学平衡.尽管目前野生型胺脱氢酶底物谱小,大多只能结合α-酮酸,对于其他的酮酸类物质无催化活性,但越来越多的研究人员从氨基酸脱氢酶出发,应用理性和半理性设计方法以及蛋白质工程技术编辑蛋白质结构,优化酶的活性、稳定性、选择性等,在利用胺脱氢酶合成手性胺研究领域取得了一系列重要进展.
1.2.1 基于亮氨酸脱氢酶的胺脱氢酶
亮氨酸脱氢酶(LeuDH, EC1.4.1.9)是一种NAD+氧化还原酶,具有脱氨作用,能够催化亮氨酸转化为2-酮异己酸[17].其对L-亮氨酸和NAD+都有很高的亲和力,在50 ℃时具有高活性,20 ℃时对L-亮氨酸和NAD+催化脱氨反应的Km值分别为0.65和0.015 mmol/L[18].
Abrahamson等[12]利用亮氨酸脱氢酶的晶体结构作为支架,基于苯丙氨酸脱氢酶活性口袋中底物-氨基酸的结合模式设计和构建了全新的亮氨酸脱氢酶,反应模式如图5.其中亮氨酸脱氢酶来自Bacillussphaericus(PDB:1LEH)[17],苯丙氨酸脱氢酶来自Rhodococcussp. M4(PDB:1C1D、1BW9)[19-20].分子生物学研究和序列比对发现,新的亮氨酸脱氢酶中Lys68与底物的羰基直接作用;氨基酸突变研究发现K68、E114、N261和V291是新亮氨酸脱氢酶活性口袋中的关键位点;最佳突变体K68S/E114V/N261L/V291C对多种酮类化合物的催化活性较野生型显著提高,并且具有高立体选择性(99.8%ee)和转化率(92%).
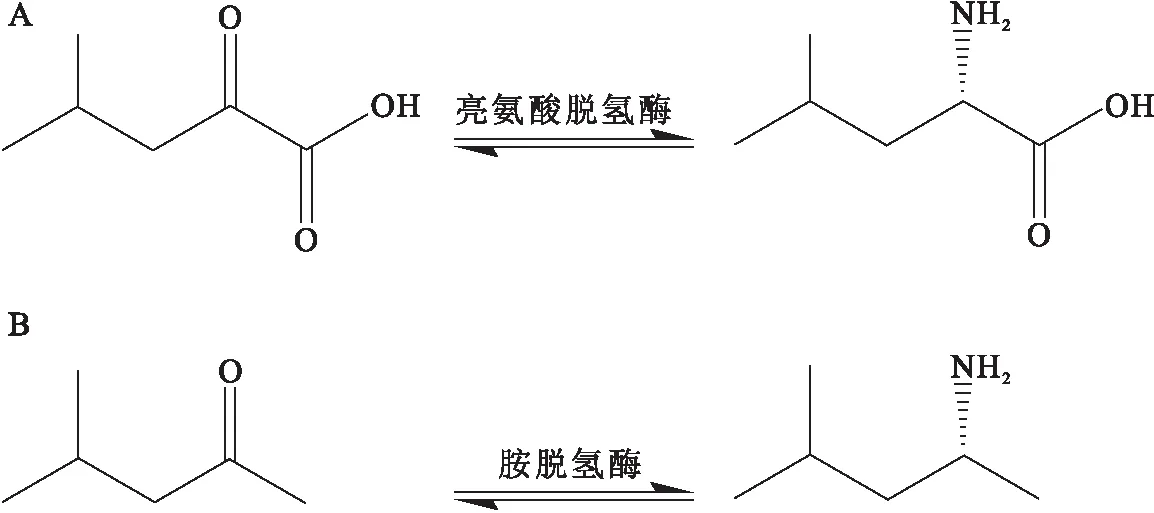
A:野生型亮氨酸脱氢酶反应;B:新型胺脱氢酶反应.
Zhou等[21]基于BcLeuDH和其他氨基酸脱氢酶序列比对以及蛋白质结构研究,对来自Bacilluscereus(ATCC 14579)的亮氨酸脱氢酶进行了改造.研究人员以ldh基因(Gene ID: 1206507)编码LeuDH.序列比对发现,在氨基酸脱氢酶中,G44、K70、K82和D117是高度保守位点,其中,G44可以与G43构成活性位点,K70能够与底物的羧基结合,K82可以增加水分子的亲和性.在酶催化过程中,底物通道对催化反应至关重要,该研究发现,Ala115、Glu116、Val118和Phe142位于底物通道中,而E116位于底物通道以及底物结合口袋的入口处,影响底物和辅酶的进入.突变后,E116V对α-酮丁酸的活力较野生型提高23%,其kcat/Km值是野生型的6倍,同时对苯乙醛酸和带脂肪族侧链的α-酮酸的催化活力也有了明显的提高.将底物结合位点的T45突变为T45M,改变了b5折叠的刚性,使底物与口袋结合更为紧密.最佳突变体T45M/E116V较野生型菌株更加稳定,活性更高.
胺脱氢酶对脂肪酮类物质具有良好的胺化活性,但是对于短链底物活性不高.对胺脱氢酶活性中心的研究发现,对脂肪族酮类物质有活性的胺脱氢酶均有相同的突变,即K68S/N261L.Chen等[22]发现,EsAmDH、LfAmDH和BspAmDH 3种酶进行 K68S/N261L突变之后,都获得了一定的催化活性,其中LfAmDH具有更长的半衰期和更高的热稳定性.LfAmDH对含有4个或5个骨架碳原子的脂肪酮有很高的还原胺化作用,但是对大的脂肪族酮化合物如2-庚酮、2-辛酮等基本没有活力.为扩大胺脱氢酶的底物范围,研究者基于亮氨酸脱氢酶(PDB:1LEH)的晶体结构创建了LfAmDH的同源模型.A113和T134是底物与活性口袋结合的关键位点,其中T134与活性口袋末端的结构有关.将A113突变为A113G时,酶对2-庚酮的活力增加;最佳突变体A113G/T134G(LfAmDH-M3)的活性口袋明显增大,与底物结合更为紧密(图6).LfAmDH-M3不仅对短链酮分子具有高活性,对分子骨架中含有6个以上碳原子的脂肪酮催化效率更是显著提高,并且具有良好的立体选择性(ee>99%).

A:LfAmDH与2-庚酮的结合位点;B:LfAmDH-M3突变体与2-庚酮的结合位点
1.2.2 基于苯丙氨酸脱氢酶的胺脱氢酶
苯丙氨酸脱氢酶的作用是将L-苯丙氨酸氧化脱氨生成苯丙酮酸,不具有将脂肪族和苄基酮类物质转化为手性胺的反应活性.Abrahamson等[23]以苯丙氨酸脱氢酶为基础构建的胺脱氢酶具有催化活性高、底物范围广的特点.突变体K77S/N276L对氟苯基丙酮(PFPA)、苯氧基-2-丙酮、2-己酮、甲基异丁基酮和3-甲基-2-丁酮等甲基酮表现出高活力(反应模式如图7所示),其中对PFPA的对映选择性超过99.8%,转化率高于90%.
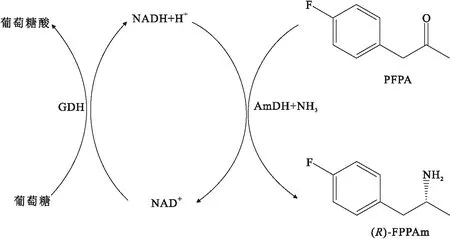
图7 基于苯丙氨酸脱氢酶改造的胺脱氢酶不对称合成(R)-1-(4-氟苯基)-2-丙胺
Knaus等[24]同样基于苯丙氨酸脱氢酶构建了活性胺脱氢酶Bb-PhAmDH.在胺脱氢酶反应中涉及辅酶循环,辅酶的活性也同样重要.研究者发现辅酶在酸性条件下很稳定,在碱性环境中失活较快.在pH为8.8时,辅酶虽有失活,但程度较低,能够满足Bb-PhAmDH生成手性胺的反应,也符合甲酸铵缓冲液的pH值范围,很好地解决了辅酶失活的问题.同时,研究发现Bb-PhAmDH对对氟苯基酮有很高的催化活性,其48 h内的转化率达到93%,立体选择性大于99%.此外,Bb-PhAmDH还可以催化其他取代类型的苯基酮,如图8所示.

图8 Bb-PhAmDH底物类型
1.2.3 基于赖氨酸脱氢酶的胺脱氢酶
赖氨酸脱氢酶(LysEDH)首次在Agrobacteriumtumefaciens中发现,在L-赖氨酸的代谢中发挥重要作用[25].野生的赖氨酸脱氢酶具有很好的热稳定性及氧化脱氨作用,只能催化L-赖氨酸的氧化脱氨反应.目前所发现的两种类型的赖氨酸脱氢酶中,L-赖氨酸6-脱氢酶(EC 1.4.1.18)催化L-赖氨酸的ε-氨基的氧化脱氨反应[26],L-赖氨酸2-脱氢酶(EC 1.4.1.15)催化L-赖氨酸的α-氨基的氧化脱氨反应[27](图9).Tseliou等[28]通过结构分析发现H181、Y238、T240、R242与α氨基和α羧基的结合相关,为底物的结合创造亲水环境;F173、V172和V130为底物的结合构建了疏水环境并迫使底物以一个正确构象与酶结合;V172、V130和 R242可能与底物的结合有关.LysEDH突变体的底物范围扩大,对图10中的底物产生活力,能够催化合成相应的手性胺,其中LE-AmDH-v1(F173A)是最佳突变体,对除6、7以外的几种酮类物质均有活力,且表现出高热稳定性.

图9 两种赖氨酸脱氢酶反应路线图[27]

图10 LysEDH突变体的酮类底物
1.2.4 胺脱氢酶小结
胺脱氢酶法制备手性胺是一种绿色高效的合成途径,但目前已发现的胺脱氢酶种类较少,且仅对α/β位不带羧基的酮表现出还原胺化作用,底物范围狭窄大大限制了其广泛应用.研究人员以氨基酸脱氢酶晶体结构为基础,通过理性设计成功构建了一系列具有广底物谱的胺脱氢酶,为手性胺的生物合成提供了思路,即可以基于已有的多种氨基酸脱氢酶的三维结构,根据反应底物的结构特征,从催化反应机理出发对酶进行理性设计,不断优化突变体的酶学性质,从而得到理想的胺脱氢酶.
2 总结与展望
现代医药和农业生产领域对手性胺化合物的需求日益增长,因此其高效制备具有广泛的应用前景和巨大的经济价值.生物酶法合成手性胺以其反应条件温和、底物价格低廉和手性选择性高等优势而受到越来越多的关注.本文综述了基于亮氨酸脱氢酶、苯丙氨酸脱氢酶、赖氨酸脱氢酶改造的胺脱氢酶在手性胺生物合成中的应用,阐述了如何利用级联反应实现辅酶循环并推动反应持续向手性胺合成方向移动的机制,同时详细探究了研究人员如何从自然存在的氨基酸脱氢酶的三维结构特征及其与底物的结合模式出发,对氨基酸脱氢酶进行理性设计和蛋白质工程改造并最终成功构建理想的胺脱氢酶的思路历程,为新型胺脱氢酶的开发指明了方向.需要指出的是,在这些合成路线中,尚存在诸多问题亟待解决,如级联反应中辅酶的快速循环、最适pH值和最适反应温度差异等.未来是绿色环保的世界,酶法必将成为主流的手性胺合成方式,生产工艺领域也必然走向多酶级联反应路线,通过“一锅”反应更快速、更高效、更优质地合成手性胺化合物.
猜你喜欢
分子催化(2022年1期)2022-11-02
中老年保健(2022年4期)2022-08-22
科学导报(2022年41期)2022-07-13
湖南大学学报·自然科学版(2020年2期)2020-04-17
贵州大学学报(自然科学版)(2020年1期)2020-02-06
首都体育学院学报(2019年5期)2019-10-18
华声文萃(2019年4期)2019-09-10
文萃报·周二版(2019年10期)2019-09-10
分析化学(2018年4期)2018-11-02
山东工业技术(2018年17期)2018-10-27
