
淫羊藿不同部位有效成分含量比较研究
2021-06-16 01:44邓寒霜杨文怡
陕西农业科学 2021年4期
邓寒霜,杨文怡,贺 博,李 月
(1.商洛学院 生物医药与食品工程学院,陕西 商洛 726000;2.陕西秦岭特色生物资源产业技术研究院,陕西 商洛 726000;3.陕西天士力植物药业有限公司,陕西 商洛 726000)
中药淫羊藿具有悠久的应用历史,具有补肝肾、袪风湿、强筋骨等功效,在我国最早的药学专著《神农本草经》中就有收录,列为上品[1~3]。我国是淫羊藿属植物地理分布中心,分布有40余种淫羊藿,2005年版《中华人民共和国药典》(以下简称"药典")收载淫羊藿药材的来源有5种小檗科植物,分别是淫羊藿(EpimediumbrevicornuMaxim)、朝鲜淫羊藿(EpimediumkoreanumNakai)、箭叶淫羊藿(Epimediumsagittatum(Sieb.et Zucc) Maxim)、柔毛淫羊藿(EpimediumpubescensMaxim)以及巫山淫羊藿(EpimediumwushanenseT. S. Ying)的干燥地上部分[4]。从2010年起,巫山淫羊藿因为与其他几种来源的淫羊藿药材在化学成分上有较大的区别,而被单独作为一味中药收录于药典中。自此,药典关于淫羊藿药材的植物来源开始减少成4种,分别是淫羊藿、箭叶淫羊藿、柔毛淫羊藿、朝鲜淫羊藿;同时,将淫羊藿药用部位由"干燥地上部分"变更为"干燥叶"[5]。至目前最新发行的2020年版药典,淫羊藿药材来源一直沿用2010年的相关规定[6,7]。为验证淫羊藿药用部位划分的合理性,比较淫羊藿植物不同部位中药用成分含量的差异,以期为扩大淫羊藿药材来源,减少自然资源的浪费提供依据,本文对来自淫羊藿不同部位的样品材料中几种有效成分的含量进行了对比分析。
1 材料与方法
1.1 材料与设备
淫羊藿原植物,采集自陕西、甘肃等地,由商洛学院执业药师李筱鉴定为小檗科植物淫羊藿(EpimediumbrevicornuMaxim)或柔毛淫羊藿(EpimediumpubescensMaxim),样品来源详细信息见表1。淫羊藿苷对照品(批号201516),购自中国药品生物制品检验所;朝藿定A(批号1804027)、朝藿定B(批号19071403)、朝藿定C(批号190625)、宝藿苷Ⅰ(批号1804026)等对照品购自北京世纪奥科生物技术有限公司。
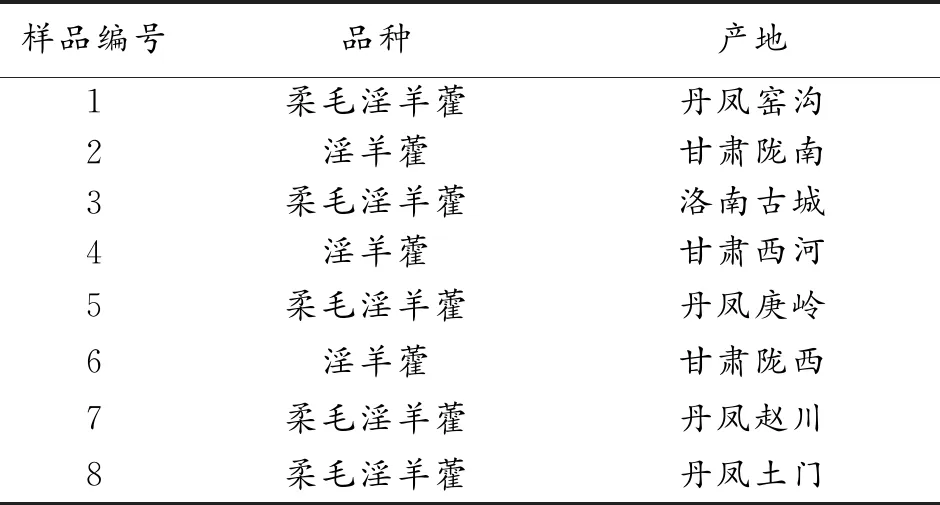
表1 淫羊藿样品产地信息
检测使用的仪器为日本岛津公司出品的LC-20A型HPLC仪,配有在线脱气机、高压泵、四元低压梯度混合装置、二极管阵列检测器、原装自动进样器、原装柱温箱、岛津LabSolutions色谱工作站等。
1.2 方法
1.2.1 样品处理 将淫羊藿原植物除去杂质,按根、茎、叶柄、叶片等不同部位分成4类供试品,干燥后粉碎,过3号药典筛(24目,筛孔内径355 μm±13 μm),即得供试品粉末。
1.2.2 含量测定 色谱条件 采用十八烷基键合的硅胶色谱柱(250 mm×4.6 mm,填料粒度5 μm),以乙腈(A)、水(B)为流动相,按表2中规定进行梯度洗脱,检测波长270 nm,柱温30 ℃,进样量10 μL,记录60 min内色谱流出曲线。
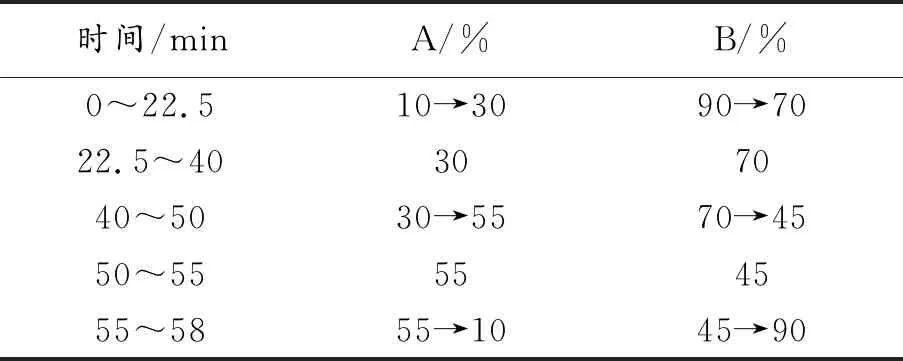
表2 淫羊藿样品含量测定梯度洗脱顺序
供试品溶液的制备 取粉碎后通过三号药典筛的样品0.2 g左右,用万分之一的电子天平准确称量样品的质量,将样品粉末装于具塞磨口瓶中,加入20 mL稀乙醇作为提取溶剂后,立即塞密,称取并记录其重量,采用超声波提取法提取样品(功率400 W,频率50 kHz),提取时间为1 h 。提取完成后,取出样品溶液放冷,再次带塞称取瓶中提取溶液的重量与前述记录比较,补充损失的稀乙醇,将样品提取溶液摇匀后,用微孔滤膜过滤,取续滤液,即得供试品溶液[7-9]。
对照品溶液的制备 取朝藿定A、B、C,宝藿苷Ⅰ以及淫羊藿苷等5种待测组分的对照品适量,准确称取其质量,用甲醇作为溶剂溶解并转移至体积为25 mL的容量瓶内,再以甲醇为溶剂定容至刻度,即得对照品溶液。
2 结果与分析
2.1 系统适应性考察结果
2.1.1 精密度考察结果 准确量取上述对照品的溶液10 μL注入HPLC仪进行分析,记录各组分色谱峰的峰面积,连续进样5次以上,经计算后得到淫羊藿苷,朝藿定A、B、C和宝藿苷Ⅰ 等5个组分各自色谱峰峰面积的RSD值分别为0.21%、0.32%、0.24%、0.31%、0.35%,考察结果证明了仪器具有良好的精密度。
2.1.2 重复性考察结果 利用同一个淫羊藿药材的样品粉末,依据前述的方法处理样品成为其供试品溶液,重复处理样品溶液5份以上,准确量取该供试品溶液10 μL注入HPLC仪进行分析,记录各组分色谱峰的峰面积,经计算后得到淫羊藿苷,朝藿定A、B、C和宝藿苷Ⅰ 等5个组分各自色谱峰峰面积的RSD值分别为0.83%、0.87%、0.74%、0.86%、1.01%,表明研究所用供试品制备方法具有良好的重复性。
2.1.3 稳定性考察结果 取同一供试品溶液,照1.2.2项中色谱条件,分别间隔0 h、4 h、8 h、12 h、24 h后,准确量取该供试品溶液10 μL注入HPLC仪进行分析,记录各组分色谱峰的峰面积,经计算后得到淫羊藿苷,朝藿定A、B、C和宝藿苷Ⅰ 等5个组分各自色谱峰峰面积的RSD值分别为2.03%、3.46%、2.81%、3.27%、3.79%,稳定性考察试验结果证明供试品的溶液其内在品质在24 h内不会发生较大变化。
2.1.4 校准曲线绘制 分别吸取淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、宝藿苷Ⅰ 不同浓度的对照品溶液进样分析,记录色谱数据,以各有效成分的浓度为横坐标(X),以峰面积为纵坐标(Y),绘制校准曲线,得各对照品曲线方程如下。
淫羊藿苷:Y=22 990 302.7778X-35 033.3333,R2=0.9999,线性范围0~0.36 mg·mL-1。
朝藿定A:Y=17 058 760.7143X+3 577.9524,R2=0.9996,线性范围0~0.32 mg·mL-1。
朝藿定B:Y=19 818 696.4286X-17 005.0000,R2=0.9999,线性范围0~0.28 mg·mL-1。
朝藿定C:Y=17 023 549.2611X+24 658.8571,R2=0.9991,线性范围0~0.29 mg·mL-1。
宝藿苷Ⅰ :Y=29 157 312.4286X+20 219.6640,R2=0.9998,线性范围0~0.28 mg·mL-1。
各对照品校准曲线见图1,混合对照品及空白对照色谱图见图2。

图1 淫羊藿样品含量测定用对照品校准曲线
2.2 含量测定结果
淫羊藿药材不同部位HPLC分析色谱图见图2,淫羊藿药材不同部位有效成分含量测定结果表3、图3。
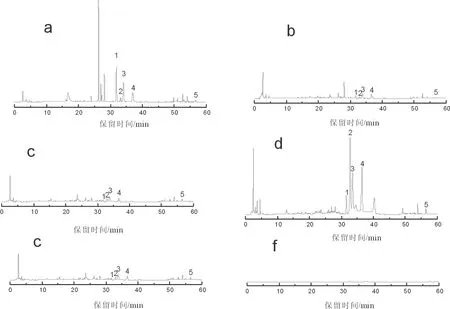
a.淫羊藿根; b.淫羊藿茎; c.淫羊藿叶柄; d.淫羊藿叶片; e.混合对照品; f.空白对照.1.朝藿定A; 2.朝藿定B; 3.朝藿定C; 4.淫羊藿苷; 5.宝藿苷Ⅰ
图3 8批淫羊藿样品不同部位中5种有效成分含量平均值比较

表3 淫羊藿药材不同部位有效成分含量测定结果
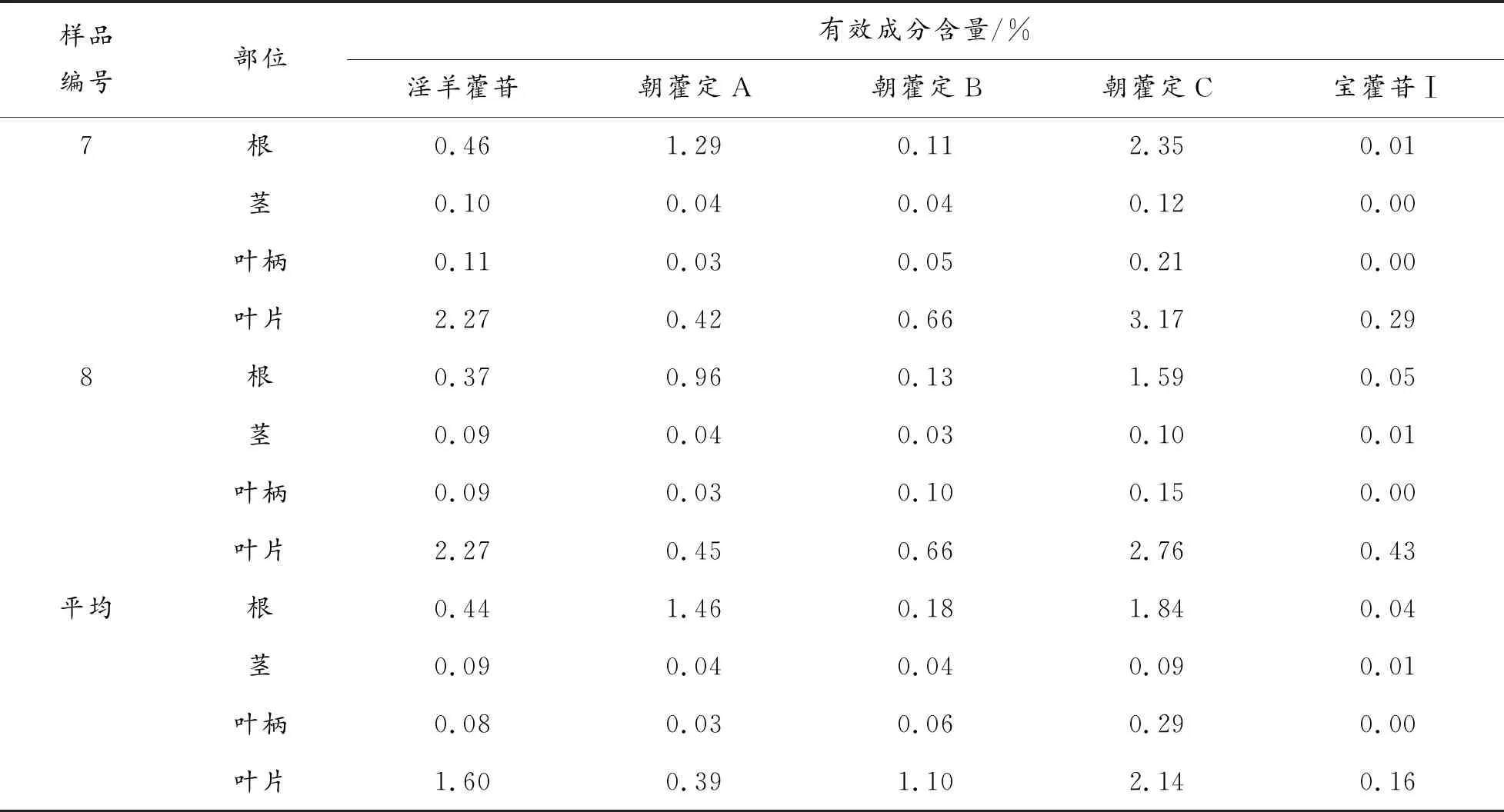
续表3 淫羊藿药材不同部位有效成分含量测定结果
由图2可知,空白对照色谱图的基线平稳,对样品分析无干扰。相较于淫羊藿根和淫羊藿叶片,淫羊藿茎、淫羊藿叶柄色谱图中色谱峰的数目少、峰面积小,表明淫羊藿此2个部位中含有的化学物质较少。淫羊藿根、淫羊藿叶片色谱图中色谱峰的数目更多、峰面积更大,表明淫羊藿根、叶片两个部位中含有更丰富的化学物质,有效成分的含量更高。淫羊藿根样品色谱图在保留时间26~28 min间,出现了3个较为明显,且在其它样品色谱图中未出现的色谱峰,表明淫羊藿根中的化学成分种类与其它部位不同,淫羊藿根与淫羊藿叶片的药理作用有区别。
由表3、图3可知,淫羊藿苷、朝藿定A、朝藿定B、朝藿定C及宝藿苷Ⅰ 5种成分在淫羊藿药材不同部位中的含量差异较为明显,其中淫羊藿叶片中有效成分含量最丰富,有4种成分的含量在各部位样品中最高,淫羊藿苷、朝藿定A、朝藿定B、朝藿定C 4种成分含量之和高于药典"淫羊藿、柔毛淫羊藿、箭叶淫羊藿均不得少于1.5%"的规定[7]。其次为淫羊藿根,朝藿定A在根中的含量最高;根中前述4种有效成分的含量之和,也能达到2020年版药典的要求;淫羊藿茎、淫羊藿叶柄中5种有效成分的含量均较低,几乎没有药用价值;宝藿苷Ⅰ的含量,在淫羊藿药材各部位中的含量均较低。上述检测结果表明,药典关于淫羊藿药用部位为"干燥叶",且生品药材中不检测宝藿苷Ⅰ的相关规定具有一定的合理性。
3 结论与讨论
研究中发现以2020版药典淫羊藿项目下规定的色谱条件,未能使朝藿定A、朝藿定B、朝藿定C以及淫羊藿苷等4种成分获得良好分离。为解决上述问题,本文在新药典规定方法的基础之上,将样品分析的色谱条件进行了调整。在调整后的色谱条件下,上述4种成分色谱峰间的分离度均可达1.5以上,各色谱峰的理论塔板数均在2.5×104以上。
多年来,淫羊藿药材生产中一直视淫羊藿原植物的根、茎、叶柄等部位为杂质,造成了一定程度上的资源浪费,也增加了生产过程中能源及人力资源的消耗。为了验证淫羊藿药用部位划分的合理性,本研究对淫羊藿不同部位中有效成分的含量进行了对比分析。由研究结果可以看出,淫羊藿茎、叶柄中几乎不含有效成分,在生产中应作为杂质除去,以增加药材品质。但淫羊藿根中化学物质含量丰富,特别是几种药典规定的指标性成分的含量之和可以达到标准要求,表明淫羊藿根具有一定的药用开发价值。课题组在今后的工作中,将重点对淫羊藿根的药理、药效作用进行研究,探明其作为淫羊藿药材使用的可行性,为拓宽淫羊藿药用资源奠定基础。
猜你喜欢
今日农业(2022年2期)2022-11-16
承德医学院学报(2022年2期)2022-05-23
中南民族大学学报(自然科学版)(2022年2期)2022-03-10
今日农业(2021年6期)2021-06-09
科学与财富(2021年34期)2021-05-10
文萃报·周五版(2020年24期)2020-06-22
分析化学(2018年4期)2018-11-02
中国药房(2018年3期)2018-10-19
分析化学(2017年12期)2017-12-25
人民周刊(2016年11期)2016-06-30
