
Characterization of the bacterial communities associated with biofilters in two full-scale recirculating aquaculture systems*
2021-06-15 08:23YuexinMAXinDUYubinLIUTaoZHANGYueWANGSaisaiZHANG
Yuexin MA , Xin DU, Yubin LIU , Tao ZHANG , Yue WANG , Saisai ZHANG
1 Key Laboratory of Environment Controlled Aquaculture, Ministry of Education, Dalian 116023, China
2 Dalian Modern Agricultural Production Development Service Center, Dalian 116023, China
3 Dalian Tianzheng Industry Company Limited, Dalian 116011, China
Abstract Bacteria play a major role in metabolizing ammonia and other metabolites in recirculating aquaculture systems (RASs). To characterize and compare the bacterial communities in the biofilters of two full-scale RASs for the culture of puff er fish, Takifugu rubripes, at diff erent ages and densities were studied. In overall, 47 807 optimized reads of the 16S rRNA gene with V4–V5 region were obtained from four biofilm samples collected after biofilm maturation. At 97% cut-off level, these sequences were clustered into 500 operational taxonomic units, and were classified into 19 bacterial phyla and 138 genera. At the phylum level, Proteobacteria and Bacteroidetes were the most abundant, followed by Nitrospirae and Planctomycetes. At the genus level, Colwellia, Marinifilum, Oceanospirillum, Lutibacter, Winogradskyella,Pseudoalteromonas, Arcobacter, and Phaeobacter were the top members. Nitrosomonas and Nitrospira were main ammonia- and nitrite-oxidizing bacteria. Diff erences in bacterial communities at diff erent sampling dates and similarities of both biofilters were revealed in the Venn diagram and cluster analysis.Maintaining a good water quality and health of farmed fish in RASs depended on the correct management of the bacterial communities. This study provides more accurate information on the bacterial communities associated with the bifilters of both RASs.
Keyword: biofilters; bacterial community; recirculating aquaculture system; Illumina-MiSeq sequencing
1 INTRODUCTION
Recirculating aquaculture systems (RASs) have been widely applied in fish production (Sakami et al.,2012; Chen et al., 2013; Ruan et al., 2015; Huang et al., 2016; Rud et al., 2017) because RASs can minimize water requirements, discharge less of better treatable wastes and improve biosecurity (Itoi et al.,2006; Martins et al., 2010; Schreier et al., 2010;Rurangwa and Verdegem, 2015; Keuter et al., 2017).Moreover, RASs provide fish with optimal water quality through recycling thus off ering the possibility to optimize feed utilization and to achieve high productions of healthy fish in controlled conditions(Rurangwa and Verdegem, 2015). In general, biofilter is an indispensable unit for RASs to maintain their water quality, which provides a fixed medium for microbial attachment and growth. Microorganisms within a biofilm formed on the surface of media play a major role in the operation of RASs. The high load of organic matter that resulted from uneaten feeds,dead bodies, and excreta of fish is converted by a diverse array of microorganisms. Nitrogen-containing organic compounds are mineralized into ammonia by heterotrophic ammonifying bacteria, with ammonia subsequently being oxidized into nitrate by autotrophic nitrifying bacteria in a biofilter. Heterotrophic bacteria within a biofilm are superior competitors for oxygen and space with autotrophic bacteria (Léonard et al.,2002; Itoi et al., 2006; Michaud et al., 2006), which are traditionally considered to hinder the eff ective operation of the system. Nevertheless, a moderate outer layer of heterotrophic bacteria in a biofilm may have a positive eff ect on nitrification by protecting nitrifying bacteria against flow detachment and grazing (Blancheton et al., 2013). Thus, it is essential to have a comprehensive understanding on the overall bacterial community structure in RASs for system management.
The advantage for puff er fish,Takifugurubripes,cultured in a commercial RAS is that they can survive and maintain winter growth. Itoi et al. (2006)characterized the bacterial community in a laboratory scale RAS for puff er fish by clone library method.Only heterotrophic bacterial community associated with multistage biofiters was investigated in a fullscale RAS for puff er fish (Chen et al., 2013). Highthroughput sequencing is rapid and cost eff ective technology that allows for in-depth taxonomic characterization of bacterial community including uncultured bacteria (Metzker, 2010; Caporaso et al.,2012). In this study, two commercial RASs containing the same components were used for the culture of puff er fish with diff erent sizes and densities. In addition, both systems had been operated for one and two years, respectively, before the experiment. The goal of this study was to characterize and compare bacterial communities in the biofilters of this two commercial RASs using Illumina Miseq sequencing.
2 MATERIAL AND METHOD
2.1 Description of experimental recirculating aquaculture system
Two full-scale RASs (systems A and B) used in this study were operated at Dalian Tianzheng Industry Company Limited (Dalian, China). Before the experiment, systems A and B have been run for one and two years, respectively. Each RAS contained nine fish-rearing tanks with a water volume in each tank of approximately 50 m3, in which the puff er fish (A: oneyear-old, mean size 210 g, 2 000 individuals per tank;B: two-years-old, mean size 720 g, 1 000 individuals per tank) were reared and fed a natural diet (fresh fish,mainlyAmmodytespersonatus) twice a day (7:00 and 17:00) at a rate of 2%–3% body weight. Water temperature ranged from 20 °C to 21 °C and salinity from 24 to 25. The effl uent from the tanks passed through a bowed screen and an air-floating tank to remove suspended and fine solids. The water circulation rate was 300 tons per hour. A submerged aerated biofilter (13.0 m in length, 3.3 m in width, and 2.0 m in depth) containing polyethylene elastic media treated metabolic wastes, which aerated by air flotation machine. An ultraviolet light chamber unit inactivated heterotrophic and coliform bacteria. Pure oxygen was injected into the contact unit using micropore diff users, and treated water flowed back to the fish-rearing tanks by gravity (Lin et al., 2017).
2.2 Sample collection
During the production of puff er fish, the biofilm samples were collected at days of 165 (A2) and 183(A3), and 157 (B2) and 175 (B3) of the systems A and B running, respectively. There is an 8-day interval between the two systems start running and the sampling dates are the same. Three sampling sites were set every 30 curtain media, and 15 sampling sites in total. Three repetitions (each contained a mixture from five sampling sites) were pooled equally as one biofilm sample. The polyethylene elastic media samples were collected in 50-mL falcon tubes, stored in a portable cooler, transferred to the laboratory, and processed immediately. The media were gently rinsed and suspended in sterile seawater, then shaken vigorously with a vortex mixer for 20 min. Biofilm samples were obtained by the liquid phase centrifugation at 6 000 r/min for 15 min.
2.3 DNA extraction and PCR amplification
DNA was extracted from four biofilm samples using a soil DNA isolation kit ( Tiangen Biochemical Technology Limited, Beijing, China) according to the manufacturer’s protocols. The 16S rRNA gene V4–V5 region was amplified by polymerase chain reaction(PCR) with primers 515F (5'-GTGCCAGCMGCCGCGG-3') and 907R (5'-CCGTCAATTCMTTTRAGTTT-3') (Turner et al., 1999) using the PCR program described by Ma et al. (2019) except for annealing at 54 °C. PCR reactions were performed in triplicate and the reaction volume was 20 μL containing 5 μL of 10× Extaq Buff er, 1.6 μL of 2.5 mmol/L dNTPs (each nucleotide), 0.8 μL of 5 μmol/L primer (each direction), 0.5 U of Extaq Polymerase, 0.3 μL of bovine serum albumin and 10 ng of template DNA.
2.4 Illumina MiSeq sequencing
Purified amplicons were obtained, pooled, and paired-end sequenced (2×300) on an Illumina MiSeq platform following standard protocols (Sun et al.,2016) (Majorbio Bio-Pharm Technology Company Limited, Shanghai, China).

Fig.1 Rarefaction curves of operational taxonomic units clustered at 97% similarity among biofilm samples of biofiter 1 (days 165 (A2) and 183 (A3)) and biofiter 2 (days 157 (B2) and 175 (B3))

Table 1 Alpha diversity based on operational taxonomic units of each biofilm sample of biofiter 1 (days 165(A2) and 183 (A3)) and biofiter 2 (days 157 (B2)and 175 (B3))
2.5 Bioinformatics analysis
Sequencing data were processed according to methods and protocols described by Sun et al. (2016)and Ma et al. (2019).
3 RESULT
3.1 Alpha diversity of the bacterial community
A total of 47 807 optimized reads with an average length of 394 bp were recovered from the four samples and clustered into 500 operational taxonomic units(OTUs). The numbers of OTUs in the A2, A3, B2, and B3 samples were 432, 376, 376, and 423, respectively.Rarefaction curves generated at the OTU level approached saturation (Fig.1), indicating that the sequencing depth was suffi cient to reflect the diversity of the bacterial community. Good’s coverage estimations revealed that 98.8%−99.7% of the species were retrieved. The richness and diversity of the bacterial community were similar among four samples on the basis of the Chao, Ace, and Shannon indices (Table 1).
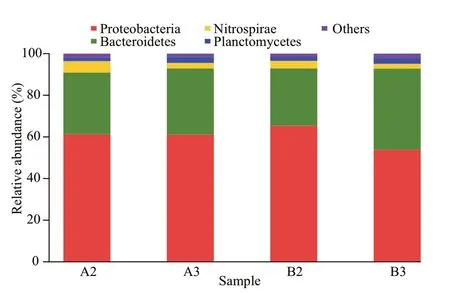
Fig.2 Taxonomic classification of bacterial reads retrieved from DNA amplicons from biofilm samples of biofiter 1 (days 165 (A2) and 183 (A3)) and biofiter 2 (days 157 (B2) and 175 (B3)) into phylum level using the RDP Classifier
3.2 Community composition
The 47 807 optimized reads were classified as 19 phyla, with Proteobacteria and Bacteroidetes making up >90% of the reads among four samples. The most abundant 16S rRNA gene sequences belonged to Proteobacteria, followed by those to Bacteroidetes(Fig.2). Relative abundances for the16S rRNA gene sequences assigned to Nitrospirae and Planctomycetes were 2.4%–5.5% and 1.7%–2.8%, respectively(Fig.2). Percentages of other phyla in four samples were less than 1% (Fig.2).
Thirty-seven classes were identified among four samples, and reads from Gammaproteobacteria and Flavobacteriia were abundant (Fig.3). The relative abundances for sequences fell within the Bacteroidia in A2, B2, and B3 samples were 12%, 11%, and 12%, respectively, while in A3 sample 4.3% (Fig.3).The percentages of sequences assigned to Alphaproteobacteria in A3 (12%) and B3 (12%)samples were higher than those in A2 (9.0%) and B2(6.1%) samples (Fig.3). The relative abundances of Nitrospira in A2, A3, B2, and B3 samples were 5.5%, 2.7%, 3.7%, and 2.4%, respectively (Fig.3).The percentages of reads from Epsilonproteobacteria were higher in A2 and B2 samples (4.9% and 5.2%)than the other two samples (<1%) (Fig.3).
One hundred and thirty-eight genera were identified in total and the sequences belonging toColwelliawere the most abundant among four samples (Fig.4). Reads fromMarinifilumwere second most abundant in A2,B2, and B3 samples, followed by those belonging toNitrospira(5.5%) andArcobacter(4.9%);Arcobacter(5.2%) andNitrospira(3.7%); andLutibacter(5.1%)andWinogradskyella(3.3%) in A2, B2 and B3 samples,respectively (Fig.4). However, sequences assigned toOceanospirillum(6.7%) were the second most abundant in the A3 sample, followed by those belonging toLutibacter(6.5%),Winogradskyella(5.7%),Pseudoalteromonas(5.3%), andPhaeobacter(3.6%)(Fig.4). In addition, reads fromVibrio(0.54%, 0.73%,0.52%, and 0.14%) andCoxiella(0.079%, 0.038%, 0,and 0.18%) were observed among A2, A3, B2, and B3 samples.
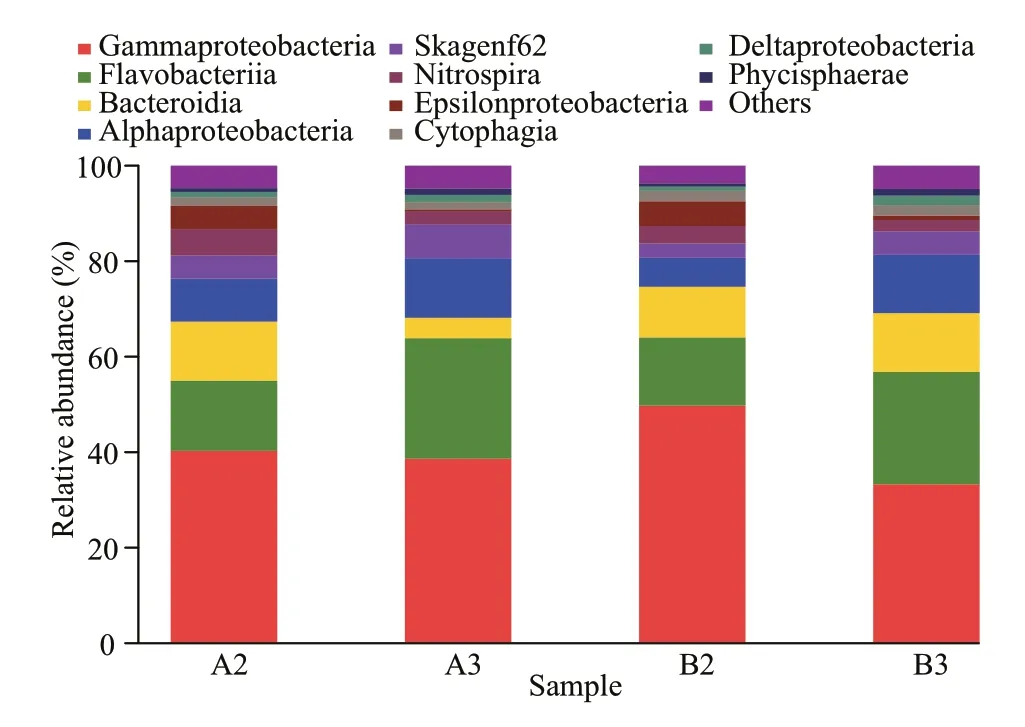
Fig.3 Taxonomic classification of bacterial reads retrieved from DNA amplicons from biofilm samples of biofiter 1 (days 165 (A2) and 183 (A3)) and biofiter 2 (days 157 (B2) and 175 (B3)) into class level using the RDP Classifier
Regarding ammonia- and nitrite-oxidizing bacteria in A2, A3, B2, and B3 samples (Fig.5), OTU 415 was identified asNitrosomonasand OTU 113 was related toNitrosococcus(Fig.5), three OTUs (numbers 132,145, and 239) were classified asNitrospira, of which OTU 145 belonged to CandidatusNitrospirasalsa(Fig.5).
3.3 Comparison of the bacterial community structure
The Venn diagram (Fig.6) indicated that 54% of OTUs and 70%–71% of OTUs were shared by four and two (A2 and A3, B2 and B3) biofilm samples.Each sample contained only a minority of unique OTUs (1–24). Variability among four samples was captured by cluster analysis (Fig.7). The results from a cluster analysis of the OTUs profile show that the A2 and B2 samples formed one cluster and the A3 and B3 samples formed another.
4 DISCUSSION

Fig.4 Taxonomic classification of bacterial reads retrieved from DNA amplicons from biofilm samples of biofiter 1 (days 165(A2) and 183 (A3)) and biofiter 2 (days 157 (B2) and 175 (B3)) into genus level using the RDP Classifier
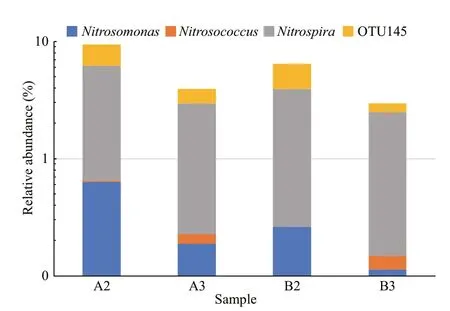
Fig.5 Relative abundances of ammonia- and nitriteoxidizing bacteria among biofilm samples of biofiter 1 (days 165 (A2) and 183 (A3)) and biofiter 2 (days 157 (B2) and 175 (B3))

Fig.6 Venn diagram showing operational taxonomic units unique to and shared by biofilm samples of biofiter 1(days 165 (A2) and 183 (A3)) and biofiter 2 (days 157(B2) and 175 (B3))
In the present study, a total of 19 diff erent phyla were identified, while the culturable bacterial community in biofilms from biofilters in another commercial puff er fish RAS were only classified into four phyla (Proteobacteria, Bacteroidetes, Firmicutes,and Actinobacteria) (Chen et al., 2013). Among the total of reads detected, Proteobacteria and Bacteroidetes were dominant phyla, which are in line with previous studies (Michaud et al., 2009; Ruan et al., 2015; Huang et al., 2016). Both phyla play an important role in degradation of organic compounds in aquatic environments (Kirchman, 2002; Kersters et al., 2006). The most abundant class in Proteobacteria was Gammaproteobacteria, followed by Alphaproteobacteria. The dominant class in Bacteroidetes was Flavobacteria. These findings are similar to previous results from a full-scale marine RAS biofilter (Ruan et al., 2015). The relative abundance of phylum Nitrospirae (class Nitrospira)(2.4%–5.5%) was in the range of other marine RAS biofilters (Ruan et al., 2015; Huang et al., 2016).Sequences related to the Planctomycetales were detected in four samples. This could suggest the presence of anammox through which nitrogen was eliminated by combining ammonia and nitrite to form nitrogen gas (van de Graaf et al., 1995). Tal et al.(2006) first confirmed the presence of anammox Planctomycetales in the microbial biofilm from the denitrifying biofilters of a RAS for the gilthead seabream,Sparusaurata. Moreover, Planctomycetes is frequently observed in biofilters oflaboratory scale and full-scale marine RASs (Itoi et al., 2006; Michaud et al., 2009; Ruan et al., 2015; Huang et al., 2016).
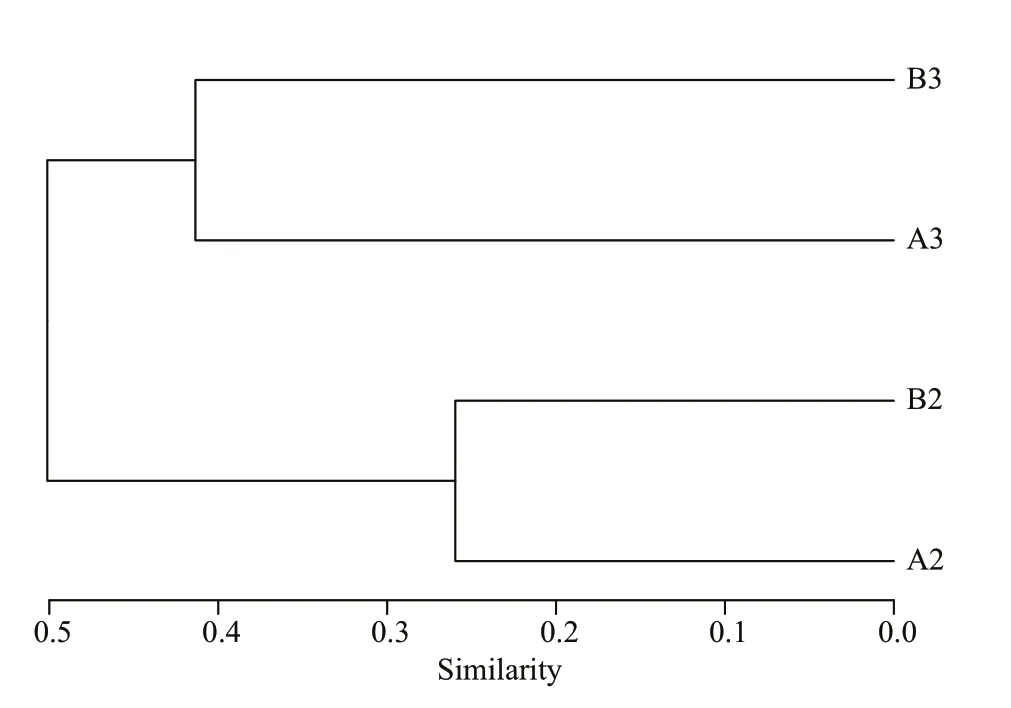
Fig.7 Cluster analysis of operational taxonomic units profile for biofilm samples of biofiter 1 (days 165 (A2)and 183 (A3)) and biofiter 2 (days 157 (B2) and 175(B3)) according to Bray-Curtis distance
At the genus level,Colwellia,Marinifilum,Oceanospirillum,Lutibacter,WinogradskyellaNitrospira,Pseudoalteromonas,Arcobacter, andPhaeobacterwere the top nine genera when data from four samples were considered together. They are diff erent from those of other puff er fish RASs ( Itoi et al., 2006; Chen et al., 2013). Using clone library method, bacteria in the aquarium (pebbles as medium)of puff er fish were composed predominantly by genera belonging toAlgibacter,Flavobacterium,Muricauda,Psychroserpens,Lewinella,Caminicella,Lactococcus,andMarinobacter(Itoi et al., 2006). Using culturedependent technique, the abundant genera wereVibrio,ShewanellaandPseudoalteromonasin biofilters(plastic brush packing medium) in a commercial puff er fish RAS (Chen et al., 2013). The diff erences in bacterial genera between puff er fish RASs may relate to fish density, biofilter construction, media, time of biofilm formation except for various methods of bacterial community analysis.Colwellia,ArcobacterandLutibacterhave been identified as abundant genera in diff erent sections of a biofilter in a RAS for full-scale tongue sole, Cynoglossussemilaevis(Ruan et al., 2015). The genusOceanospirillumwas the second most abundant in A3 sample, members of this genus are known to be distributed ubiquitously in marine environments (Satomi et al., 2002). This taxon can utilize amino acids or the salts of organic acids as carbon sources (Pot and Gillis, 2015). In addition,PseudoalteromonasandShewanellawere also isolated from the intestine of puff er fish (Li et al., 2015), which might be the source of bacteria for the biofilters.
In line with previous studies (Tal et al., 2003; Foesel et al., 2008; Ruan et al., 2015; Huang et al., 2016), it was demonstrated that representatives of the genusNitrospirawere identified as dominant nitrite-oxidizing bacteria (NOB) in biofilters of marine RASs. The relative abundance of genusNitrospiraacross the four samples ranged from 2.4% to 5.5%, which included a marine nitrite-oxidizingNitrospiraspecies CandidatusNitrospirasalsa(Haaijer et al., 2013). In addition, the sequence number of the NOB was roughly an order of magnitude higher than that of ammonia-oxidizing bacteria, which included generaNitrosomonasandNitrosococcus, suggesting that the NOB may not be the limiting factor in nitrification performance. These findings indicate thatNitrosomonasandNitrospiraplay an important role in removing ammonia and nitrite from puff er fish RASs. Similar results were observed in biofilters of other marine RASs (Ruan et al., 2015;Huang et al., 2016).
Several potential pathogens, such asCoxiellasp.andVibriosp., were detected in this study.Vibriospp.have been isolated from biofilms of biofilters in another commercial puff erfish RAS (Chen et al.,2013).Coxiellasp. andVibriosp. have been observed associated with the biofilters packing medium in a RAS for sea bass,Dicentrarchuslabrax(Michaud et al., 2009). Members of both genera have also been found in biofilters among several marine RASs(Huang et al., 2016). Despite the presence of potential pathogens, no diseased fish were observed during the period of this study. One of reasons might be that the relative abundances of the potential bacterial pathogens in biofilters of puff er fish RASs were low(Coxiella, 0–0.18%;Vibrio, 0.14%–0.73%).
The results obtained of this study suggest that both biofilters contain a large bacterial diversity, with similarities in the bacterial communities being observed between the two systems as shown in the cluster analysis. Although the puff er fish sizes and densities were diff erent in both systems, the daily feeding dose for two-years-old puff er fish was slightly less than that for one-year-old, thus the loads of organic metabolic wastes and ammonia on both systems at the same sampling date were not much diff erent. However, there were diff erent bacterial communities at the two sampling dates, this might be explained by the bacterial decomposition of substrate,making the nutrient substances of bacteria diff erent at diff erent sampling dates.
5 CONCLUSION
Proteobacteria and Bacteroidetes were the most abundant phyla in both biofilters that involved in the degradation of organic compounds.NitrosomonasandNitrospirawere main ammonia- and nitriteoxidizing bacteria being responsible for ammonia and nitrite oxidation. Both biofilters harbored similar bacterial communities and dissimilar at the two diff erent sampling dates. These results are useful for the management and stable operation of the puff er fish RASs.
6 DATA AVAILABILITY STATEMENT
All data generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
 Journal of Oceanology and Limnology2021年3期
Journal of Oceanology and Limnology2021年3期
- Journal of Oceanology and Limnology的其它文章
- Steady increase in water clarity in Jiaozhou Bay in the Yellow Sea from 2000 to 2018: Observations from MODIS*
- Phylogenetic diversity and bioactivity of culturable deepsea-derived fungi from Okinawa Trough*
- Allelopathic eff ects of mixotrophic dinoflagellate Akashiwo sanguinea on co-occurring phytoplankton: the significance of nutritional ecology*
- Investigation of the decline of Ulva prolifera in the Subei Shoal and Qingdao based on physiological changes*
- Effi ciency of phosphorus accumulation by plankton,periphyton developed on submerged artificial substrata and metaphyton: in-situ observation in two shallow ponds*
- Petroleum exploitation enriches the sulfonamide resistance gene sul2 in off shore sediments