
Construction of high-density genetic linkage map of Pyropia yezoensis (Bangiales, Rhodophyta) and identification of red color trait QTLs in the thalli*
2021-06-15 08:22LuWANGKuipengXUXianghaiTANGJunhaoWANGFannaKONGYunxiangMAO
Lu WANG, Kuipeng XU , Xianghai TANG, Junhao WANG , Fanna KONG ,**,Yunxiang MAO
1 Key Laboratory of Marine Genetics and Breeding(Ministry of Education), College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China
2 Key Laboratory of Utilization and Conservation of Tropical Marine Bioresource(Ministry of Education), College of Fisheries and Life Science, Hainan Tropical Ocean University, Sanya 572022, China
3 Marine Biology and Biotechnology Laboratory, Pilot National Laboratory for Marine Science and Technology(Qingdao),Qingdao 266071, China
Abstract Pyropia yezoensis is an important macroalga because ofits extensive global distribution and economic importance. Color is an important trait in the thalli of P. yezoensis, it is also an eff ective marker to identify the hybridization in genetic breeding. In this study, a high-density genetic linkage map was constructed based on high-throughput single nucleotide polymorphism (SNP) markers, and used for analyzing the quantitative trait loci (QTLs) of red color trait in the thalli of P. yezoensis. The conchospore undergoes meiosis to develop into an ordered tetrad, and each cell has a haploid phenotype and can grow into a single individual.Based on this theory, F1 haploid population was used as the mapping population. The map included 531 SNP markers, 394.57 cM long on average distance of 0.74 cM. Collinear analysis of the genetic linkage map and the physical map indicated that the coverage between the two maps was 79.42%. Furthermore, QTL mapping identified six QTLs for the chromosomal regions associated with the red color trait of the thalli.The value of phenotypic variance explained (PVE) by an individual QTL ranged from 4.71%–63.11%. And QTL qRed- 1- 1, with a PVE of 63.11%, was considered the major QTL. Thus, these data may provide a platform for gene and QTL fine mapping, and marker-assisted breeding in P. yezoensis in the future.
Keyword: Pyropia yezoensis; high-density genetic linkage map; quantitative trait loci (QTL) mapping; F1 haploid population; red pigment variant
1 INTRODUCTION
The genusPyropia/Porphyra(Bangiales,Rhodophyta), popularly known as nori and laver, is one of the most economically important seaweeds in the world (Sutherland et al., 2011). According to FAO’s statistics, in 2017, the global nori production was about 2.56 million tons in fresh weight, with a commercial value of more than $2.3 billion (http://www.fao.org/fishery/factsheets/en). In China,PyropiayezoensisandPyropiahaitanensisare the two most commercially valuablePyropiaspecies.P.haitanensisis mainly cultivated in the Southern coastline of China.P.yezoensisis mainly cultivated in the Northern coastline of China and also cultivated in Korea and Japan. The life cycle ofP.yezoensisconsists of two alternative phases, macroscopic leafy thallus and microscopic filamentous conchocelis. The conchocelis, also called the sporophyte, is diploid and can release conchospores, which undergo meiosis and germinate into haploid thalli. It has been reported thatP.yezoensishas the chromosome numbers of 2n=6(Yabu and Tokida, 1963; Migita, 1967; Yabu, 1969)and the nuclear genome size is fewer than 100 Mb(Nakamura et al., 2013).
The selection and breeding of new varieties is important for developing economical seaweeds for the seaweed industry. The cultivation and breeding of laver originated in the 1950s (Fei et al., 1998).Traditional breeding oflaver in China was based on selection, mutation, and crossing (Hu, 2006). Many chemical and physical techniques including application of chemical mutagens (Yan et al., 2000, 2004), γ rays(Wang et al., 2000; Yan et al., 2005), and heavy-ion beam (Niwa et al., 2011) was used to produce mutants ofPyropia, most of them were pigment mutations. In recent years, crossbreeding has been widely performed to produce new varieties with new traits. For example,Pyropiasp. from India was crossed withP.haitanensisto produce an improved strain (HR-5), which was characterized by fast growth, high quality, and production oflarge amount of conchospores (Ding et al., 2018). The intraspecific hybridization of the greentype pigmentation mutant and red-type pigmentation mutant ofP.yezoensiswas carried out, and then, an improved strain (A-18) with fast growth rate and wildtype color was isolated from the recombinants (Jiang et al., 2018). However, these traditional methods were limiting because of the time-consuming, ineffi cient,and diffi cult to distinguish phenotype for the blade plasticity for environmental factors. They were the bottlenecks in the development oflaver industry. With the development of molecular breeding, molecular marker-assisted selection (MAS), and whole genome selection breeding, which have become the main tools providing direction for plant breeding enabling plant breeders to directly select the genotypes, thereby accelerating crop improvement (Johnson, 2003;Moose and Mumm, 2008; Ribaut et al., 2010; Yu et al.,2011; Qing et al., 2019; Randhawa et al., 2019; Wang et al., 2019). High resolution genetic linkage map has been the basis for these breeding strategies.
Genetic linkage maps have been essential for the genetics studies, genomic evolution, and mapping quantitative trait locus (QTL) (Cheng et al., 2019).To date, genetic linkage maps have been successively constructed for dozens of diff erent species of plants and animals. In many crops, genetic linkage maps were also used to identify the QTLs associated with economic traits, for example plant height (PH), leaf length (LL), maizechloroticmottlevirus(MCMV)resistance, flag leaf-related traits, grain shape and salinity tolerance (De Leon et al., 2016; Liu et al.,2018; Chen et al., 2019; Choi et al., 2019; Sitonik et al., 2019). As compared to the land crops, the development of genetic linkage maps in seaweeds is relatively lacking. The genetic linkage maps have been reported for only few species of seaweeds. A tentative amplified fragment length polymorphismsimple sequence repeat (AFLP-SSR) linkage map ofLaminariawas constructed with an average distance of 7.91 cM (Yang et al., 2009). InSaccharinajaponica, based on the specific length amplified fragment sequencing (SLAF-seq), 7 627 single nucleotide polymorphism (SNP) loci were mapped to 31 linkage groups with an average spacing of 0.69 cM, and the QTLs for the blade length and blade width phenotypes were detected (Wang et al., 2018b).A genetic map ofEctocarpussiliculosuswas constructed containing 406 AFLP markers resulting in 34 linkage groups, which spanned a total distance of 2 459.3 cM and an average distance of 6.6 cM(Heesch et al., 2010). InUndariapinnatifida, a highdensity genetic linkage map was constructed using SLAF-seq technique, which spanned a distance of 1 816.28 cM and consisted of 30 linkage groups,with an average distance of 0.39 cM (Shan et al.,2015). InPyropia, the first high-density genetic linkage map was constructed inP.haitanensisusing SLAF-seq, which included 4 550 length polymorphic markers. These markers were combined into 740 bins on five linkage groups, with a length of 874.33 cM and an average distance of 1.18 cM between the adjacent bins (Xu et al., 2015). Recently, a genetic map ofP.yezoensiswas constructed using only 92 sequence-related amplified polymorphism (SRAP)loci to produce three linkage groups (LGs), with a mean interlocus space of 6.23 cM (Huang and Yan,2019).
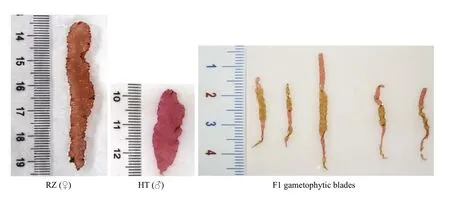
Fig.1 The thallus of the male parental strain (RZ), the female parental strain (HT), and the F1 thallus selected from the cross between the RZ and HT strains
Although two genetic linkage maps ofPyropiahave been constructed, some problems still exist.Thus, it is still necessary to construct a genetic linkage map ofPyropiawith high quality and density,especially forP.yezoensis. It was diffi cult to apply the molecular markers technique in laver breeding because the thallus of F1 gametophyte was chimera.The color mutants of the thalli were widespread used in the genetic studies ofPyropia/Porphyra(Yan et al., 2000). In nature or mutant breeding ofP.yezoensis, it is common to identify red pigment variants or mutants. A red pigment variant was found previously in the laboratory, and the contents of phycoerythrin and phycocyanin were significantly higher than that in the wild type. And we found that the red color trait probably was a qualitative trait,which could be used to distinguish genotypes of the tetrad. So far, there is no research on the red color trait of thalli yet. Therefore, in order to apply this red color trait as a marker in molecular pyramiding breeding, the QTL mapping for the red color trait was imperative. In the present study, we constructed a high-density genetic linkage map ofP.yezoensisusing SNP markers identified by genome resequencing and F1 haploid population, and further detected the red color trait of thalli using the QTL method. Our study has provided a means to correlate the traits and genotypes inP.yezoensisand may contribute towards the ultimate goal of molecular marker-assisted breeding.
2 MATERIAL AND METHOD
2.1 Plant material
Two pure lines, RZ and HT, with diff erent blade colors were used as the parent strains to construct the mapping populations in this study. Pure RZ line with a wild-type, established by our laboratory was the maternal parent (Fig.1). Single somatic cells were enzymatically isolated from the thallus of RZ under the microscope with glass capillaries. Pure HT line with a red-type was the paternal parent and a spontaneous mutant which was isolated from a pure line PYL-349 (Fig.1). Healthy and immature thalli of parents were separately cultured in 500-mL flasks containing PES medium (UTEX Culture Collection of Algae) and agitated by aeration at 15 °C under 80 μmol photons/(m2·s) (10-h L:14-h D) (Niwa, 2010).
2.2 Construction of map population
The crossing was performed according to the method of Niwa (2010). The diagrammatic sketch is shown in Fig.2. Two pure lines, HT line and RZ line,were used for the cross to construct the mapping population. The selected male and female thalli were co-cultured at a ratio of 3:1 for about 10 days until carposporangia appeared in the maternal piece. Then the maternal blade pieces were washed and gently rubbed in a petri dish with sterilized seawater which were both filtered and boiled. The cleaned pieces were chopped with a blade, then, the carpospores were sucked out with glass micropipettes (inside diameter, 0.5 mm) and cultured in 96-well plates to grow into F1 conchocelis colonies at 20 °C under 30 μmol photons/(m2·s) (12-h L:12-h D). When the conchocelis colonies grew to a certain size, they were fragmented to inoculate the shells, and continually cultured at 24 °C under 30 μmol photons/(m2·s) (10-h L:14-h D) for about a month and a half until the conchosporangia formed. Once the conchospores were released and attached to the cotton strings at 20±1 °C under 135 μmol photons/(m2·s) (12-h L:12-h D), they were transferred to a flask containing 800 mL of PES culture medium and cultured with aeration at 20±1 °C under 135 μmol photons/(m2·s)(12-h L:12-h D) to develop into thalli. The culture medium was renewed every 3 days. After 2 weeks,the temperature was reduced to 10±1 °C and the culture was continued for one and a half months under the conditions of 160 μmol photons/(m2·s) (12-h L:12-h D) until the thalli were obtained. The F1 thalli with only four color-sectors were selected and each color-sector blade was singly developed into an individual. These thalli were obtained to form the F1 haploid population.
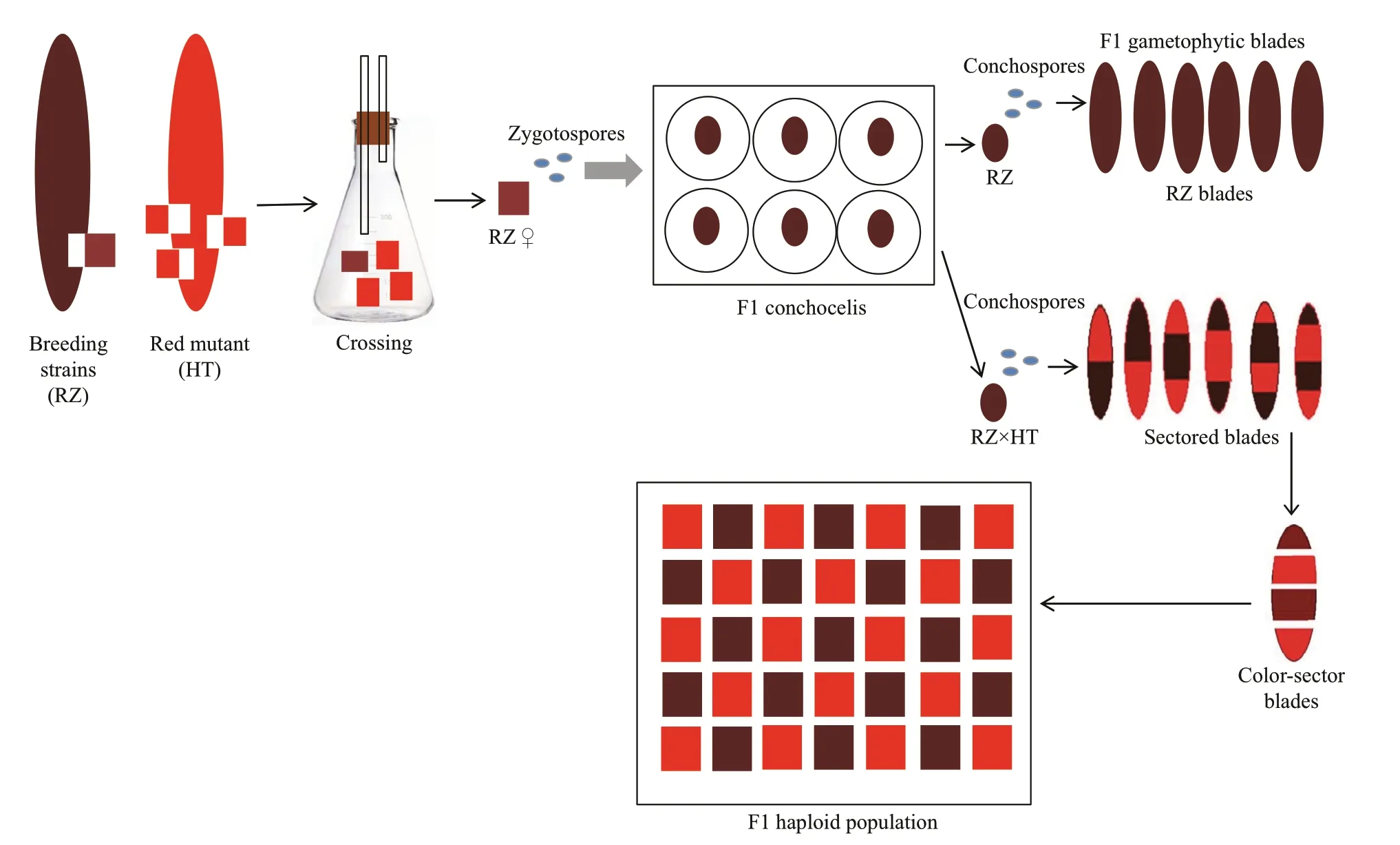
Fig.2 Schematic view of the cross between RZ and HT, and construction of F1 haploid population of P. yezoensis
2.3 DNA extraction and Illumina library construction
DNA was isolated from the thallus of each parental line and F1 haploid lines. The collected blades were ground into powder in liquid nitrogen, and the DNA was extracted and purified using the Plant Genomic DNA Kit (DP305-02, TIANGEN) following the manufacturer’s method. The DNA quality was determined using a Nanophotometer N60 (Implen,Germany) and the concentration was determined based on the Qubit2.0. The size and integrity of DNA were assessed by 1.0% agarose gel electrophoresis using lambda DNA as the standard. After qualified detection, DNA samples were used for Illumina library construction using the VAHTS Universal DNA Library Prep Kit for Illumina®V3 ND607 (Vazyme Biotech Co., Ltd; Nanjing, China) according to the manufacturer’s protocol.
2.4 High-throughput sequencing and SNP genotyping
The eligible libraries were diluted for pair-end sequencing on Illumina Hiseq™ Xten sequencing platform (Illumina, Inc; SanDiego, CA, USA) at Novogene Bioinformatics Technology Co. Ltd. in Tianjin, China. Data quality control was performed based on the obtained resequencing data considering the following four criteria: i) removal of adapter contaminated reads; ii) the low quality (bases with a mass value less than 5 and accounting for more than 20% of the total reads) reads were dropped; iii)removal of reads with a high ratio of N (the base measured as N accounts for more than 10% of the total reads); and iv) avoiding duplication.
The clean reads of the parents and F1 individuals were first mapped to the reference genome(WMLA00000000) with BWA backtrack (Li and Durbin, 2009). The reference genome was indexed and the command ‘aln’ was used with the parameters‘- o 1 - e 10 - t 4 - l 32 - i 15 - q 10’ to find the suffi x array coordinates of good matches for each read (Xu et al., 2019). The best alignments were then converted into the Sequence Alignment/Map (SAM) format using the ‘sample’ command (Li and Durbin, 2009).We improved the accuracy of sequence alignment using the following filtering criteria: i) removal of the number of mismatched bases less than 5 and the alignment result with a quality of 0; ii) use of Picard software (McKenna et al., 2010) with the command‘AddOrReplaceReadGroups’ to get a BAM file, and used command ‘FixMateInformation’ to ensure each read and its mate pair were synchronized; iii) Removal of potential PCR duplicates.
After alignment, used the SAMtools program (Li et al., 2009) with Bayesian algorithm, SNP calling was performed on a population scale. The command‘mpileup’ was used with the parameters ‘- q 1 - C 50 - S- D - m 2 - F 0.002 - u’ (Xu et al., 2019). The subsequent analysis filtered the raw SNPs using Variant Call Format (VCF) tools (Danecek et al., 2011) and(Genome Analysis Toolkit) GATK (McKenna et al.,2010) using the following criteria: i) coverage depth≥ 4, ≤ 1 000; ii) mapping quality ≥ 20; iii) distance of adjacent SNPs ≥ 5 bp; iv) retention of only those SNPs that occurred in more than 50% of the individuals; v)minor allele frequency (MAF) ≥ 0.05.
During data processing for genotyping, since the genotype of F1 individual was haploid, they were artificially doubled into diploid. The SNPs between the parents were considered as the original markers for genotyping of 131 individuals, and only the individual with the segregation patterns “aabb” were retained for genetic linkage map construction. Genotyping was defined by covering at least 80% of all individuals. It means that at least 105 individuals out of 131 individuals can be detected genotypes (a/b) at a site.
2.5 Genetic linkage map construction
The genetic linkage map was constructed by Joinmap 4.1 (Cheema and Dicks, 2009). Marker segregation ratios were calculated using the chisquare test, and the markers showing significant segregation distortion (P<0.001) were excluded.Logarithm of the odds (LOD) value ≥5.0 was used as thresholds for groups. The map distances were calculated by Kosambi mapping function. Genetic maps were drawn using lepmap3.0 software.
2.6 Evaluation of the genetic map
In our group, high-throughput chromosome conformation capture (Hi-C) technique was used to assemble the genome ofP.yezoensisat the chromosome level. Three scaff olds were obtained that were equivalent to the three chromosomes observed in haploidP.yezoensis(Ohme and Miura, 1988). The quality of the genetic map was evaluated with heat maps generated for each LG, and the collinear analysis between the genetic linkage map and the physical map was performed. The pair-wise recombination values of all the mapped SNP markers were used to assess the quality of the genetic linkage map in heat maps. Furthermore, heat maps were also to reflect the relationship of recombination between the markers from one single LG (Shan et al., 2015). Collinear analysis reflected the positional relationship of markers between the genetic linkage map and the physical map. Both drawings were implemented in R language.
2.7 Quantitative trait locus (QTL) analyses of red color trait
The phenotypes of F1 individuals for color as the criterion were statistically analyzed; the red color was denoted as R, and the normal color was denoted as W.MapQTL6.0 program was used to determine the phenotypic thresholds, and the significant LOD thresholds for the red color trait were calculated by the permutation test ofα<0.05 andn=1 000 for significant linkages (Van Ooijen and Kyazma, 2009).Based on these permutations, QTL analysis was carried out using WinQTLcart2.5 (Wang et al., 2007),and the composite interval mapping (CIM) method(Zeng, 1994) was performed to detect any significant associations between the red color trait and marker loci. A LOD score of 3.0 at genome level was used as a minimum cutoff to declare the presence of a QTL.The value of phenotypic variance explained (PVE) of a major QTL is defined as more than 15% (Kumawat et al., 2012; Liu et al., 2014; Wang et al., 2018a).
3 RESULT
3.1 The construction of F1 haploid population
Based on the scheme in Fig.2, pure line RZ (♀)was crossed with HT (♂) and carpospores were released from fertilized carposporangia on the maternal blade piece, which developed into conchocelis. Then the conchocelis colonies were further released conchospores, which grew to F1 thalli. Among the F1 thalli that were obtained, only the chimeras of color-sectors with two or three or four color-sectors (Fig.2) showed that the crossing process was successfully. In this research, only the thalli with tetrad chimera were selected for constructing population. Finally, 152 color-sectors from 38 F1 thalli were obtained as mapping population.
3.2 SNP calling and genotyping
A total of 152 color-sectors were used to build the DNA libraries, and only 131 were successful. Two parents and 131 individuals were used to generate SNP data for subsequent analysis, and 96% of the sequences were correctly aligned to the reference genome ofP.yezoensis. In total, 24.24 Gb clean data were generated for the parents, 8.81 Gb for the female parent (RZ), and 15.43 Gb for the male parent (HT).The average sequencing depth was 75.89× in RZ and 144.21× in HT. For 131 individuals, a total of 676.59 Gb clean data were obtained containing 43.57 hundred million reads, on average of 150 bp in length for each read. The number of reads per single individual ranged from 0.99–75.35 million, with an average of 33 million reads and the sequencing depth of 46.27×.
A total of 131 F1 haploid individuals from colorsectors were used to construct the mapping population.For each blade color-sector, its genotype was haploid.It could develop into double haploid conchocelis by self-cross, with a homozygous genotype. Thus, in this study, to reduce the time and to improve the effi ciency,we did not complete the self-crossing of the F1 blade color-sectors. Before genotyping, the genotypes of F1 color-sector individuals were artificially directed as doubled into diploid. Then a F1 haploid population of 131 individuals was constructed and used for genotyping and constructing the genetic linkage map.The F1 haploid population was genotyped by 24 438 SNP polymorphic markers between the two parents,and finally 15 822 polymorphic marker loci were successfully genotyped in 131 individuals(Supplementary Table S1). These markers showed the aa×bb segregation pattern. No abnormal bases were found, indicating that the accuracy of genotyping was high. After filtering out the markers with less than 80% genotypic integrity coverage, 668 markers were finally obtained. The segregation distortion thresholdpwas set to be 0.001, and only three distorted segregation markers were filtered out. Ultimately, a total of 665 valid markers were entered into the linkage analysis.
3.3 High-resolution genetic linkage map construction
After linkage analysis, 531 of the 665 markers were successfully mapped onto the genetic linkage map, while other 134 markers failed to link to any group. The 531 markers were distributed on the three linkage groups (Supplementary Table S2). The final genetic linkage map was 394.57 cM in length with an average distance of 0.74 cM between the adjacent markers (Fig.3, Table 1). The number of LGs in the genetic linkage map was same as the chromosome number of the haploid genome (3 chromosomes) ofP.yezoensis(Yabu and Tokida, 1963; Migita, 1967;Yabu, 1969). As shown in Table 1, the length of the three LGs was 176.22, 141.35, and 77.00 cM,respectively (mean, 131.52 cM). The degree of linkage between the markers was reflected by “Gaps<5 cM”, which ranged between 97.436% and 99.425%, with an average value of 98.542%. Among the three LGs, LG1 was the largest LG, including 162 SNP markers and spanning 176.22 cM with an average marker interval of 1.09 cM. The second largest group was LG2 with 195 SNP markers and spanning 141.35 cM. The average inter-marker distance for LG2 was 0.72 cM. LG3 was the most saturated and the shortest, containing 174 SNP markers spanning 77.00 cM with an average marker interval of 0.44 cM.
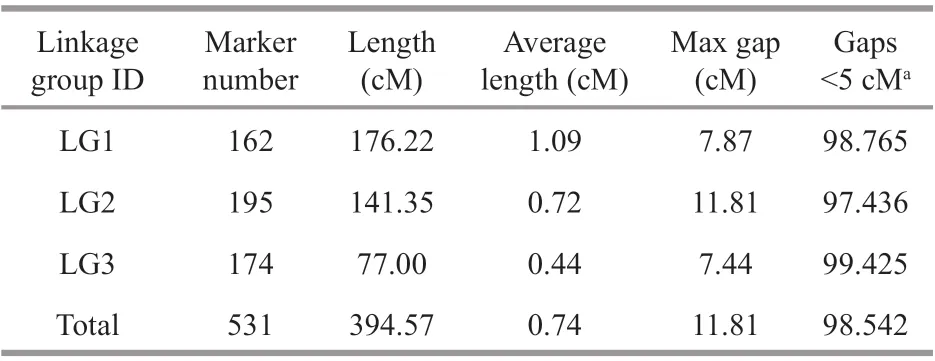
Table 1 Summary statistics of the genetic linkage map of P. yezoensis
3.4 Evaluation of the genetic map
Collinearity analysis was performed by mapping the markers on the genetic linkage map against the scaff olds, as shown in Fig.4. In general, three linkage groups showed high collinearity corresponding to the scaff olds. For LG1 and scaff old_1, the region before marker mk37 at the 120 cM could correspond to the physical region from 1 to 21 617 320 bp on scaff old_1 in turn; while an inversion occurred behind the region of 120 cM on LG1 and the region from 25 184 629 bp to the end of scaff old_1. And the inverted region in LG1 contained a total of 35 markers. For LG2 and scaff old_2, most of the marker positions except the region from 66 cM to 91 cM which contained 76 markers could correspond to scaff old_2 as well.Similarly, the marker positions of LG3 could correspond to scaff old_3 consistently. Overall, the coverage between the physical and genetic linkage maps was 79.42%.
The heat map reflects the recombination relationship between the markers from one single linkage group, which can be used to identify errors in the order. As shown in Supplementary Figs.S1–S3,the markers on each linkage group were used to calculate pair-wise recombination values using three heat maps. If the order between the markers was correct, the color on the diagonal was the darkest and reddest, indicating that the linkage relationship was the strongest. The linkage relationship between the markers became weaker as the distance increased. In general, the visualization of the heat map indicated that the LGs performed well.

Fig.3 High-density linkage map for P. yezoensis
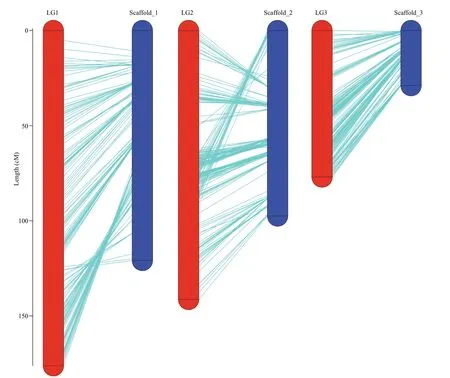
Fig.4 Collinearity analysis of genetic map and physical map in P. yezoensis
3.5 Identification of red color related QTL
The gametophytic phenotype of parent RZ is naturally brown. The paternal parent HT is the natural variant from RZ, with a red color gametophytic phenotype. The phenotypes of blade color trait, of the parents and the 131 F1 singles were recorded as the phenotype data of the QTLs (Supplementary Table S3). Among the 131 F1 singles, there were 70 wildtype (R) and 61 red-type (H) (Fig.5). The statistical significance of the QTL eff ects was evaluated by 1 000 permutations, and the threshold at genomewide significance level 0.05 of the LOD scores was determined as 3.0. According to the threshold at genome level, 6 QTLs associated with the red color trait were identified on LG1 and LG2; no QTLs were found on LG3 (Table 2). Among the 6 QTLs, the minimum and maximum LOD scores were 3.194 and 27.157 respectively, and the value of PVE ranged from 4.71%–63.11% (Table 2).
The QTLqRed-1-1was located at 49.9–50.3 cM between mk159 and mk162 on LG1, with the maximum PVE value of 63.11%. This locus explained 63.11% of the phenotypic variance, indicating that itmay be a major locus for the red color trait. Based on the genome data of this region, from 37 302 208 bp to 41 082 413 bp on scaff old_1, a total of 501 genes were identified, 282 were annotated in the nonredundant (NR) database (Supplementary Table S4)and 219 genes with unknown functions. The Gene Ontology (GO) enrichment analysis of these 282 genes showed that their functions including enzymatic activity, basal metabolic process, photomorphogenesis,and so on (Supplementary Table S5). Subsequent fine mapping and gene editing techniques are needed for further verification. The QTLqRed-1-2was located at 59.1–60.4 cM, with the physical position between 21 327 428 bp to 21 420 62 bp of scaff old_1,explaining 14.83% of the phenotypic variance. The QTLqRed-2-1was located on LG2 at position 22.6–23.6 cM, with the physical position between 32 143 876 bp to 32 430 593 bp of scaff old_2,explaining 10.22% of the phenotypic variance. The QTLqRed-2-2was located on LG2 at position 34.4–36.3 cM, with the physical position between 30 634 713 bp to 32 430 699 bp of scaff old_2,explaining 14.09% of the phenotypic variance. In addition, the other two QTLs (qRed-2-3andqRed-1-3) were located at 40.0–45.2 cM and 74.0–79.1 cM along LG2 and LG1, with PVE of 8.12% and 4.71%,respectively. The CI (95% confidence interval for QTL length) ranged from 0.4–5.1 cM among the 3 QTLs on LG1, and the CI ranged from 1.0–5.2 cM among the 3 QTLs on LG2, and half of them were less than 1.0 cM. Therefore, 3 QTLs with a distance ofless than 1.0 cM from the nearest markers could be used for further studies.
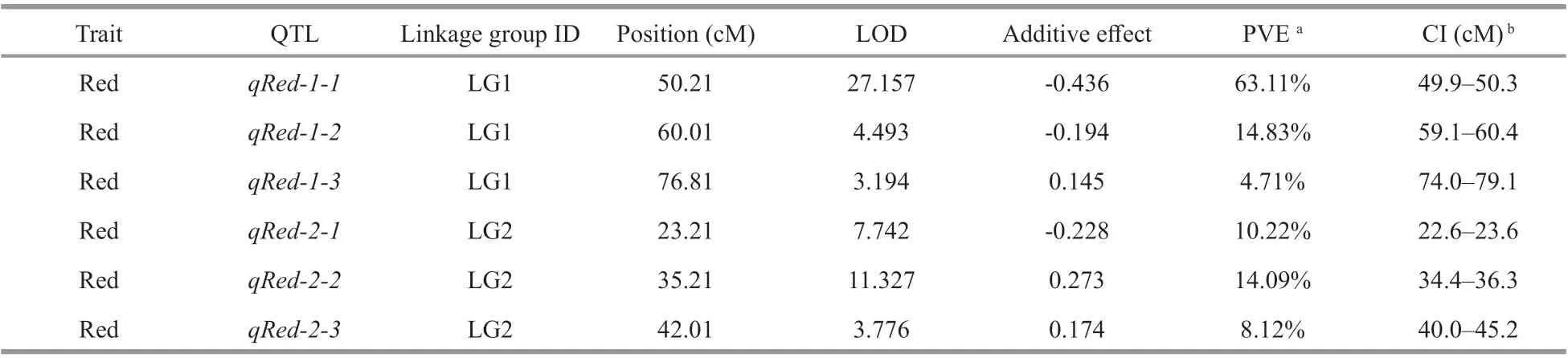
Table 2 QTLs detected for red color trait based on CIM analyses in the F1 haploid population
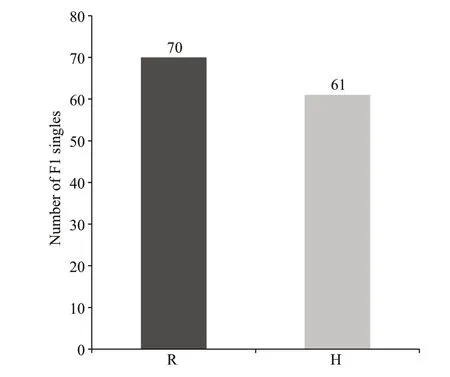
Fig.5 The distribution of blade color trait in the 131 F1 singles
4 DISCUSSION
4.1 Mapping population of P. yezoensis
The construction of population map is the first step in constructing a genetic linkage map. Currently,among the commonly used mapping populations,such as backcross 1 (BC1), F2 generation population,double haploid (DH) population, and recombinant inbred lines (RIL), the DH population is considered as the ideal mapping population because ofits homozygous genotype. In plants, there are various methods to construct DH population, such as anther culture, haploid induction, and natural doubling (Zhu et al., 2004; Chai et al., 2007; Ma et al., 2018; Shen et al., 2018). ForP.haitanensis, its gametophytic single cell in vitro can directly develop into conchocelis and the genotype can be doubled into diploid. Thus, it is easy to construct a DH population forP.haitanensisafter obtaining F1 thallus. A DH population of 100 lines was built to construct the genetic linkage map ofP.haitanensis(Xu et al., 2015). However,P.yezoensisdoes not own this character. Its thalli are monoecious and could be self-fertilized. Thus, it can complete double haploid by self-fertilization.
In addition, F1 conchospores ofPyropiadivided into ordered tetrads via meiosis, which were chimeras.If there were no obvious markers, it was diffi cult to select the heterozygous F1 thallus to ensure if the crossing occurred. The color sector embedded in the F1 heterozygous thallus is an ideal selection marker to resolve this problem (Yan and Aruga, 2000). The ordered tetrad continued to divide and develop into 2–4 pigment containing chimeras. In this study, we used red color pigment variant to make a cross with the wild type, and then F1 thalli with color-sectors were used to construct the mapping population(Fig.2).Since each color-sector of the thalli is haploid,after the thalli self-fertilized to form carpogonium and developed into conchocelis, their genotypes were doubled. Huang and Yan (2019) used this strategy to construct a genetic linkage map ofP.yezoensis, but the process was time-consuming. Conchospore germination ofP.yezoensisdeveloped in a linear pattern, and cell divisions were not synchronous (He et al., 2019). In this study, although we selected F1 haploid population as the mapping population to construct the genetic map, yet during genotyping, the genotypes of F1(a/b) individuals were automatically doubled and changed into aa/bb. We confirmed that this strategy is very scientifically feasible.
4.2 Features of the high-density map of P. yezoensis
As important macroalgae in aquaculture, a highdensity genetic linkage map is essential for molecular marker-assisted selection and QTL mapping ofP.yezoensis. The acquisition of high-density markers is critical to increase the density of genetic linkage maps. High-throughput sequencing provides support for the development of high-throughput SNP molecular markers at the whole genome level,including SLAF, Restriction-site associated DNA sequencing (RAD-seq), IIb restriction site-associated DNA (2b-RAD) and re-sequencing (Davey et al.,2011; Sun et al., 2013). As a result, reducedrepresentation genome sequencing (RRGS)technology has also emerged. Although SLAF-seq was used to develop long polymorphic markers inP.haitanensis, but most of them were found to be redundant (Xu et al., 2015). This may be due to the peculiarity ofPyropiagenome as lots of microorganisms andPyropiathalli showed serious symbiosis relationship. When isolating the genome ofPyropia, most of the nucleic acids are derived from the microorganisms. In addition, the genome of plastids and mitochondria also account for a very high ratio. Among the 1748 polymorphic markers from SLAF-seq, only two markers could be used for constructing the genetic map (Xu et al., 2015). Then,the bin-marker approach was further utilized to construct the final genetic map, but it still lacked genomic information for reference. Huang and Yan(2019) used fluorescent SRAP markers to construct the genetic linkage map ofP.yezoensis; this technique is based on PCR technology, which inevitably limits the density of the map and the throughput of the molecular markers. The map included 92 SRAP markers (Huang and Yan, 2019); however, the SRAP primers were designed from higher plants(DendrobiumloddigesiiRolfe,EucommiaulmoidesOliver andMalusMill), and most of the loci were located in the open reading frame (ORF) regions of the gene. In addition, each marker listed on their map was one of the bands of SRAP amplification products and its sequence was not provided in this paper.Therefore, it was diffi cult to determine the locations of SRAP markers on the genome and integrate our map with their map.
In this study, we applied re-sequencing based on the reference genome (WMLA00000000) with superior quality to develop SNP markers at the genomic level. All the contaminating sequences from the bacteria, plastids, and mitochondrial genome were removed from the genome sequences. Using this high-quality reference genome, more eff ective SNP markers were mapped to the genome to increase the density of the markers. As compared to the published genetic map ofPyropia, such asP.haitanensis, with an average density of 1.18 cM andP.yezoensiswith an average density of 6.23 cM (Xu et al., 2015; Huang and Yan, 2019), the mapping in our study with an average density of 0.74 cM is the highest (Fig.3,Table 1). For mapping, 668 markers out of 15 822 polymorphic markers were finally obtained based on 80% genotypic integrity coverage. After filtering out three distorted segregation markers, a total of 665 valid markers were entered into the linkage analysis.The rest of the 15 154 markers showed no recombination and were found to be redundant.Although our map has the highest density as compared with otherPyropia, the density of our map can be further improved by increasing the parental polymorphism and the number of population individuals.
High-resolution genetic mapping with many markers is helpful in improving the genome sequence assembly in plants (Ganal et al., 2011). The number of LG in our study was 3 (Table 1), which is equivalent to the haploid chromosome number ofP.yezoensis(Ohme and Miura, 1988). This indicated that the mapping strategy in this research was scientific. Most regions of the genetic linkage map showed high linearity with the scaff olds of the reference sequence ofP.yezoensis(Fig.4). Some short inversion and translocation phenomena existed in the partial region(Fig.4). Given that the reference genome comes from RZ pure line, the occurrence of the inversion and translocation may be because the genome of pure line HT was used in crossing. Thus, this phenomenon has also provided a guide to confirm and improve the assembly of genome sequence data.
4.3 QTLs of red color traits
In nature or mutant breeding, it is easy to identify color variants or mutants. Color variants or mutants play an important role in the genetic studies ofPyropia/Porphyra(Miura and Shin, 1989). Ohme et al.(1986) used the mutant pigment color as the marker to clarify meiosis as the dividing process for the conchospore. Ohme and Miura (1988) also used tetrad analysis to determine the distance between the centromere and the loci of four color mutants that were assigned to three diff erent linkage groups. Color variants or mutants have also been used as genetic markers for intraspecific or interspecific crossbreeding ofPyropiato produce improved lines (Yan and Aruga,2000; Liu et al., 2015). Although color mutants have also been used to construct genetic linkage maps(Huang and Yan, 2019), there are no reports regarding their genetic location on the chromosomes.
We first identified 6 QTLs distributed on 3 LGs that associated with red color trait, including one major QTL and five minor QTLs (Table 2). The PVE of a major QTL is normally defined as more than 15%(Kumawat et al., 2012; Liu et al., 2014; Wang et al.,2018a). Therefore, in this study, the QTLs with PVE larger than 15% were defined as the major QTLs. It was noted that major QTLqRed-1-1showed a PVE of up to 63.11%, with the highest LOD value of 27.157 and the 95% CI of 49.9–50.3 cM (Table 2). The PVE of the remaining five QTLs were all less than 15%.Therefore, we believe that the contribution of these five minor QTLs to red color trait could be negligible,and QTLqRed-1-1made an absolute contribution to the red color trait. Higher LOD values could better control the occurrence of false QTLs and are suitable for fine mapping of the major QTLs (Li et al., 2010;Tao et al., 2013). Thus, QTLqRed-1-1should be further analyzed to identify the genes controlling the red color trait.
The color-related traits in higher plants showed that they were controlled by the single-gene or QTL regions through QTL analysis. The monogenic mutations lead to changes in the fruit color of pepper(Brand et al., 2012). The red color leaf of peach and the yellow color ofLophopyrumponticumare also controlled by single gene, theGrgene, and the yellow pigment geneY, respectively (Dirlewanger et al.,2004; Zhang et al., 2005). Yet, the leaf color in Chinese cabbage (BrassicarapaL. subsp.pekinensis)and switchgrass (Panicumvirgatum) have been detected to be controlled by the QTLs (Xu et al.,2007; Lowry et al., 2015). In pot azalea (Rhododendronsimsiihybrids), pink petal color has been confirmed to result from a gene-dosage eff ect, and two major QTLs were found to be associated with the petal color(De Keyser et al., 2013). Candidate genes involved in skin and flesh coloration in peach (PprMYB10),cherry (PavMYB10), and apple (MdMYB1/MdMYBA/MdMYB10) have also been reported (Kui et al.,2010; Sooriyapathirana et al., 2010; Frett et al., 2014).In the present study, the major QTL for the red color trait of blade,qRed-1-1on LG1, existed on scaff old_1 from 37 302 208 bp to 41 082 413 bp (Supplementary Table S2), and a total of 501 genes were identified.According to the NR functional annotation, 282 genes were annotated with known functions (Supplementary Table S4). GO enrich analysis showed that the genes related to organelle organization accounted for the largest rate, and the genes related to catalytic activity and cellular components ranked second. To prove the authenticity of a QTL, it is necessary to convert a QTL from a statistical to a genetic trait (Mo, 2003).Further reducing the distances between the QTLs with red color trait by fine-mapping is necessary to identify the genes controlling this trait. The gene knock-out or genome editing technologies can also be used to validate the genes controlling the red color trait ofPyropia.
5 CONCLUSION
In this study, a F1 haploid mapping population consisting of 131 individuals was constructed based on the color-sectors F1 thallus from the cross between the pure line RZ and the red variant HT. By genome resquencing, a high-density genetic linkage map was constructed, containing three linkage groups with a total length of 394.57 cM and an average genetic distance of 0.74 cM. This map showed better collearity with the scaff old ofP.yezoensisgenerally. Six QTLs related to blade red color trait were mapped to linkage groups 1 and 2. QTLqRed-1-1with PVE of 63.11%was considered as the major QTL. These results will not only facilitate marker-assisted selection breeding ofP.yezoensis, but also lay a foundation for further isolating of the genes control red color trait.
6 DATA AVAILABILITY STATEMENT
The data generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
 Journal of Oceanology and Limnology2021年3期
Journal of Oceanology and Limnology2021年3期
- Journal of Oceanology and Limnology的其它文章
- Steady increase in water clarity in Jiaozhou Bay in the Yellow Sea from 2000 to 2018: Observations from MODIS*
- Phylogenetic diversity and bioactivity of culturable deepsea-derived fungi from Okinawa Trough*
- Allelopathic eff ects of mixotrophic dinoflagellate Akashiwo sanguinea on co-occurring phytoplankton: the significance of nutritional ecology*
- Investigation of the decline of Ulva prolifera in the Subei Shoal and Qingdao based on physiological changes*
- Effi ciency of phosphorus accumulation by plankton,periphyton developed on submerged artificial substrata and metaphyton: in-situ observation in two shallow ponds*
- Petroleum exploitation enriches the sulfonamide resistance gene sul2 in off shore sediments