
Molecular identification and population diff erentiation of Aurelia spp. ephyrae in sea cucumber aquaculture ponds of northern China*
2021-06-15 08:10SaijunPENGQingqingLIULeiWANGTingtingSUNTamaraSHIGANOVAZhijunDONG
Saijun PENG, Qingqing LIU, Lei WANG, Tingting SUN , Tamara SHIGANOVA ,Zhijun DONG
1 Muping Coastal Environment Research Station, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China
2 University of Chinese Academy of Sciences, Beijing 100049, China
3 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China
4 Shirshov Institute of Oceanology Russian Academy of Sciences, Moscow 117997 Nakhimovskyi pr., 36, Russia
Abstract Aurelia spp. ephyrae have been reported to form blooms in sea cucumber aquaculture ponds in the Bohai and Yellow Seas. To identify the species, we carried out a genetic analysis of Aurelia spp. ephyrae and medusae based on mitochondrial 16S rRNA gene. Samples of four Aurelia sp. ephyrae populations were collected in sea cucumber aquaculture ponds and samples of four Aurelia sp. medusae populations were collected in coastal waters. Using a BLASTn search, we found that both the ephyrae collected in the aquaculture ponds and medusae collected in coastal waters belong to Aurelia coerulea. Seventeen haplotypes were recovered from the 16S rRNA gene. The overall haplotype diversity and nucleotide diversity of the 166 A. coerulea individuals were 0.686% and 0.329%, respectively, indicating high haplotype diversity and low nucleotide diversity. Moreover, the haplotype diversity of ephyrae populations were generally lower than that of medusae populations with close sampling points. The genetic diff erentiation between ephyrae populations collected in the sea cucumber aquaculture ponds and A. coerulea medusae collected in coastal waters was not significant, suggesting the ephyrae populations in the sea cucumber culture ponds were part of the same genetic group as the medusae populations in the coastal waters. Phylogeographic analysis of the 16S rRNA region revealed that there was no significant correlation between the haplotypes and the geographic distribution of populations. Pairwise fixation index values showed significant genetic diff erentiation and limited gene flow between A. coerulea population of Weifang and other locations.
Keyword: Aurelia coerulea; medusae; ephyrae; 16S rRNA gene analyzes; genetic diff erentiation; genetic variability
1 INTRODUCTION
Jellyfish blooms have become frequent in the last few decades as a result of global climate change and human activities that have led to local ecological shifts and had harmful eff ects on biodiversity and commercial stocks (Arai, 2001; Mills, 2001; Purcell,2005; Uye, 2008; Richardson et al., 2009; Falkenhaug,2014). Representatives of genusAureliaare among the most common species that form jellyfish blooms.There have been many reports ofAureliaspecies blooms across the world, including in China, Japan,Korea, Spain, and Tunisia (Toyokawa et al., 2000;Dong et al., 2010; Baxter et al., 2011; Uye, 2011;Wang et al., 2012; Purcell et al., 2013; Bosch-Belmar,et al., 2016). The genusAureliabelongs to Cnidaria,Scyphozoa, Semaeostomeae, Ulmaridae and is considered the most common genus of scyphozoan jellyfish worldwide (Lucas, 2001; Chinese Zoology Editorial Committee, Chinese Academy of Sciences,2002; Schroth et al., 2005).Aureliaspp. have a metagenetic life cycle with an asexual benthic generation and sexual planktonic medusae(meroplanktonic species) (Lucas, 2001).
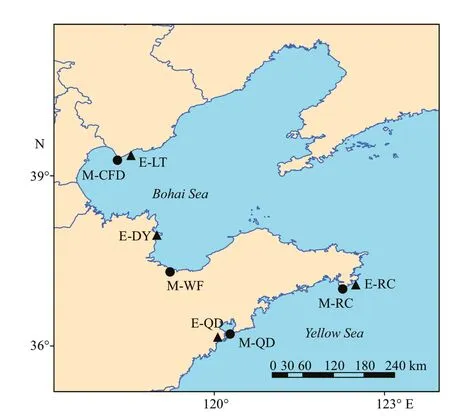
Fig.1 Sampling sites of Aurelia spp. in Chinese coastal waters
Many researchers believed in the past that genusAureliaconsisted of 3 valid species (Aureliaaurita,Aurelialimbata, andAurelialabiata) and 10 cryptic species (Aureliasp.1–Aureliasp.10) globally(Dawson and Jacobs, 2001; Schroth et al., 2002;Dawson, 2003, 2005; Ki et al., 2008). However,Scorrano et al. (2017) showed thatAureliasp.1(Aureliacoerulea) andAureliasp.8 (Aureliasolida)can be considered valid species, whileAureliasp.5(Aureliarelictasp. nov.) must be described as a new species. There are geographical diff erences in distribution between species. For example,A.coerulea(Aureliasp.1) occurs mainly in warmtemperate regions, including China, Australia,California, France, Japan, and Korea, whileAureliasp.2 occurs mainly in Brazil;Aureliasp.3 and sp.6 in Palau;Aureliasp.4 in Indonesia, Palau, and Hawaii;A.relictasp. nov. (Aureliasp.5) in the Mljet Lake of Croatia;Aureliasp.7 in New Zealand and Tasmania;A.solida(Aureliasp.8) in the Northern Adriatic Sea and the Gulf of Lyon;Aureliasp.9 in the Gulf of Mexico; andAureliasp.10 in Alaska and European seas (Dawson and Jacobs, 2001; Schroth et al., 2002;Dawson, 2003, 2005; Ki et al., 2008; Ramšak et al.,2012; Dong et al., 2015; Scorrano et al., 2017). From 2003 to 2014,Aureliaspp. medusae and ephyrae blooms frequently occurred in the Yellow Sea and Bohai Sea in China, including in aquaculture ponds,water power plants, docks, and so on. In coastal sea cucumber culture ponds, the typical blooms ofAureliaspp. ephyrae have been observed. The local farmers call them “red jellyfish” because during blooms they cause the surface of the aquaculture ponds to appear red.
Mitochondrial DNA is widely used in the study of species population genetic analysis due to its simple structure, high base mutation rate, rapid evolution rate, high sensitivity, maternal inheritance, and general lack of recombination (Brown et al., 1979;Whitmore et al., 1994; Hallerman, 2003).Mitochondrial 16S ribosomal RNA (rRNA) is a very commonly used molecular marker. Dong et al. (2017)determined by phylogenetic analyses of 16S rRNA that the “red jellyfish” ephyrae, which appeared in the culture ponds of the Rongcheng Shidao area in China belonged toA.coerulea. The ephyrae in aquaculture ponds is hypothesized to enter the ponds via water flow from the Yellow Sea and Bohai Sea. However,genetic analysis ofAureliaspp. medusae and ephyrae has not yet been carried out in other culture ponds.This study analyzes the genetic structure and diversity of theAureliaspp. population in sea cucumber aquaculture ponds along the coast of the Bohai and Yellow Seas based on the 16S rRNA gene. In total, 81Aureliaspp. ephyrae were collected from multiple sea cucumber culture ponds in 4 regions and 85Aureliaspp. medusae were collected from 4 coastal locations.The aim of our study was to identify species ofAureliaspp. ephyrae in sea cucumber aquaculture ponds in the Bohai and Yellow Seas. Finally, the genetic diff erentiation between ephyrae populations collected in the sea cucumber aquaculture ponds andA.coeruleamedusae collected in coastal waters were also revealed using the 16S rRNA gene.
2 MATERIAL AND METHOD
2.1 Sample collection
From 2014 to 2016, a total of 166Aureliaspp.individuals were collected in the Bohai Sea and Yellow Sea during the local jellyfish blooming periods(Supplementary Table S1; Fig.1). Of these, 85 medusae were collected across four geographic locations: Caofeidian (M-CFD), Qingdao (M-QD),Rongcheng (M-RC), and Weifang (M-WF), while 81 ephyrae were obtained fromApostichopusjaponicusaquaculture ponds in Laoting (E-LT), Qingdao (EQD), Rongcheng (E-RC), and Dongying (E-DY). The whole ephyrae and the medusae tissue extracted from the bell margin were preserved in 99% ethanol and then stored at -20 ℃ until DNA extraction.
2.2 DNA extraction, PCR amplification,sequencing, and alignment
The genomic DNA of medusae tissue was extracted using the TIANamp Marine Animals DNA Kit(TIANGEN, Beijing, China), while the genomic DNA of ephyrae was extracted using the CTAB(Cetyltrimethyl Ammonium Bromide) method. DNA from both life stages was stored at -20 °C. A region of the 16S rRNA gene was amplified using the published primers (16S-H 5′-CAT AAT TCA ACA TCG AGG-3′and 16S-L 5′-GAC TGT TTA CCA AAA ACA TA-3′)(Ender and Schierwater, 2003). Polymerase Chain Reactions (PCR) were performed in a total volume of 50 μL containing 50–100-ng genomic DNA, 1×PCR buff er, 2.5-U Taq DNA polymerase, 1.5-mmol/L MgCl2, 0.2-mmol/L dNTPs, and 0.25-mmol/L primers. The protocol for 16S rRNA amplification was as follows: 5 cycles of 4 °C for 1 min, 45 °C for 50 s and 72 °C for 1 min; 30 cycles of 94 °C for 50 s,50 °C for 1 min and 72 °C for 1 min; and a final elongation at 72 °C for 5 min. The PCR products were analyzed using electrophoresis on a 1% agarose gel, stained with GenecolourTM(Biotium, USA).
PCR products were purified and sequenced directly using ABI 3730 automated DNA sequencer at Shanghai Sangon Biological Engineering Technology& Service Co., Ltd., China. All PCR products were sequenced in both directions to obtain accurate sequences. 16S rRNA sequences were aligned using CLUSTALX 1.83, and were verified, edited and assembled with BioEdit 7.1. To ensure correct alignment, the sequences were conducted with MEGA 7.0.
2.3 Data analyses
The nucleotide composition and variation between sites were analyzed in MEGA 7.0 (Kumar et al.,2016). The haplotype diversity (Hd) and nucleotide diversity (π) were calculated using DnaSP 5.10(Librado and Rozas, 2009). Levels of overall interpopulation diff erentiation as well as diff erentiation between diff erent region populations and populationpairwise diff erentiation were estimated using Φ-statistics, which give an analogue of F-statistics calculated within the analysis of molecular variance(AMOVA) framework, calculated using the Arlequin 3.5 (Excoffi er and Lischer, 2010). A median-joining network showing the intuitive and accurate relationships between the haplotypes was constructed using the Network 4.6 (Bandelt et al., 1999). The sequences ofA.coeruleasamples in this study can be downloaded from Genbank (MF981165–MF981181,Supplementary Table S2).
3 RESULT
3.1 Species identification and genetic variability
16S rRNA sequences were obtained from 166 individuals. The aligned sequence length of the 16S rRNA sequences was 532 bp. BLASTn analysis indicated that both the ephyrae collected in the aquaculture ponds and medusae collected in coastal waters belong toA.coerulea.
Nineteen polymorphic sites were detected in 532 sites, with a mutation rate of 3.57%, including 6 parsimony information sites, accounting for 1.13%,and 13 singlet nucleotide mutation sites, accounting for 2.44%. Seventeen haplotypes were defined in 19 polymorphic sites, defined as Hap 1–17. The genetic distance between haplotypes ranged from 0.2% to 1.5%, and the average genetic distance was 0.7%. The average contents of bases A, T, C, and G were 25.7%,35.4%, 19.4%, and 19.5%, respectively. The A+T content (61.1%) was significantly greater than the C+G content (38.9%). The Hd and π of each geographic region are shown in Table 1. Across all samples, the haplotype diversity ranged from 0.448±0.134 to 0.755±0.084, while the nucleotide diversity ranged from 0.190%±0.059% to 0.421%±0.062%. Overall haplotype diversity in samples was 0.686±0.032, and the corresponding nucleotide diversity was 0.329%±0.019%, showing a higher haplotype diversity and lower nucleotide diversity. The highest haplotype diversity was found in the M-CFD population and the lowest in the M-WF population. The M-CFD population also had the highest nucleotide diversity and the E-QD population had the lowest. The haplotype diversity of ephyrae populations were generally lower than that of medusae populations with close sampling points. The Hd of E-LT was lower than M-CFD, the same results still existed in E-QD and M-QD, E-RC and M-RC.
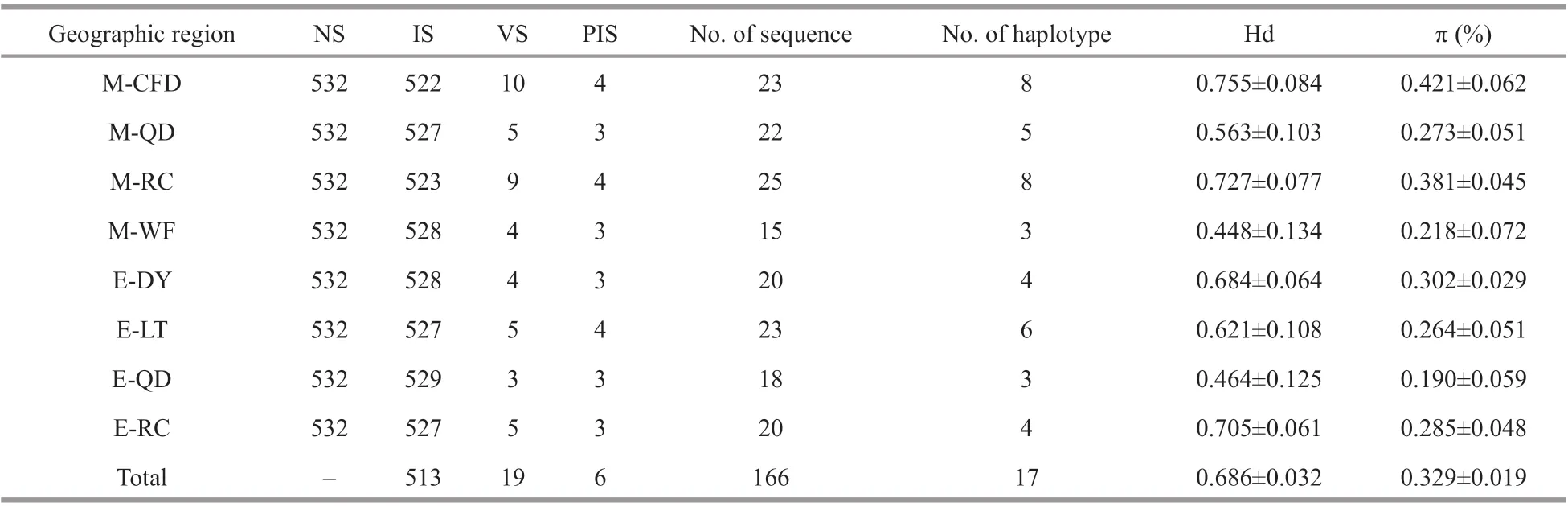
Table 1 Genetic diversity of mitochondrial 16S rRNA sequences in A. coerulea according to geographic region
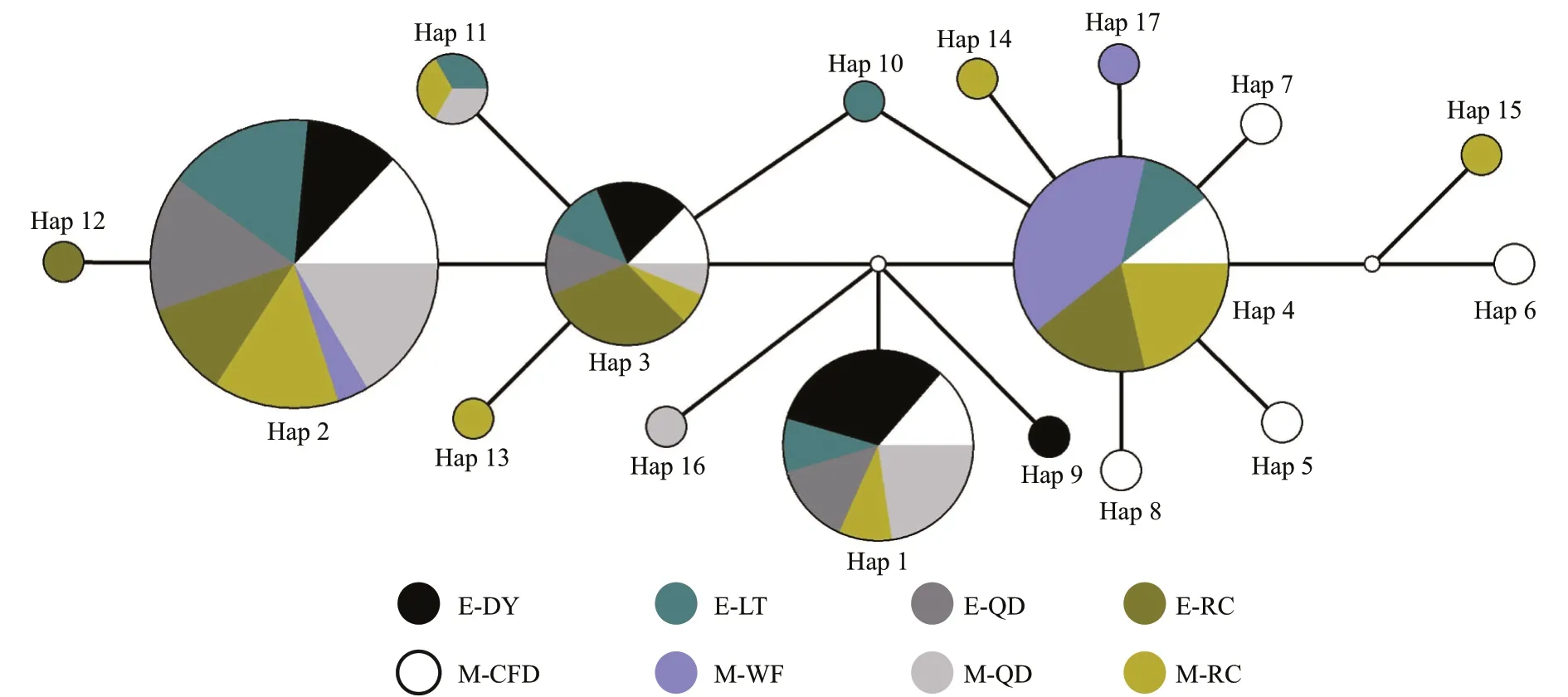
Fig.2 Median-joining networks for all A. coerulea 16S rRNA haplotypes
3.2 Population genetic diff erentiation
Based on the median-joining haplotype network method, a network relationship diagram ofA.coeruleahaplotypes was constructed (Fig.2). There were 5 haplotypes (Hap 1, Hap 2, Hap 3, Hap 4, and Hap 11) that were found in individuals from more than one geographic region. Hap 2 was the most frequent haplotype, occurring across all eight geographic regions. Hap 1 was found in the M-CFD, M-QD,E-QD, E-DY, M-RC, and E-LT populations. Hap 3 occurred in all populations except M-WF. Hap 4 was found in the M-CFD, M-RC, M-WF, E-LT, and E-RC populations and Hap 11 occurred in the M-QD,M-RC, and E-LT populations. With the exception of E-QD, other populations had their own specific haplotypes. There was no significant correlation between the haplotypes and the geographic distribution of populations.
The analysis of molecular variance (AMOVA)revealed that 89.02% of the genetic variation occurred within populations (P<0.01), whereas 13.13%occurred among regions (P<0.05). Thus, the AMOVA revealed significant genetic diff erentiation between the six regions (CFD, QD, RC, WF, DY, LT)(φCT= 0.131,P=0.025), and extremely significant genetic diff erentiation within populations in the total samples (φST= 0.110,P=0) (Table 2). However, there was no significant genetic diff erentiation among populations within regions (P=0.735).
Population-pairwiseFstvalues ranged from-0.033 5 (M-RC/M-CFD) to 0.542 3 (E-QD/M-WF).These results indicated that the E-DY, E-LT, E-RC,M-RC, E-QD, M-QD, and M-CFD populations weresignificantly diff erentiated from the M-WF population (Fstrange: 0.176 2 to 0.542 3,P<0.05).The genetic diff erentiation betweenA.coeruleamedusae collected in coastal waters and ephyrae populations collected in the sea cucumber aquaculture ponds was not significant. Moderate genetic diff erentiation existed between the M-WF population and the M-RC and M-CFD populations(0.15<Fst<0.25). Between the M-WF population and the E-DY, E-LT, E-RC, E-QD, M-QD populations,theFstvalues were all greater than 0.25, and the correspondingNmwas between 0 and 1, indicating a large genetic diff erentiation between populations(Table 3).Nm>4 indicates more frequent gene exchange between groups.
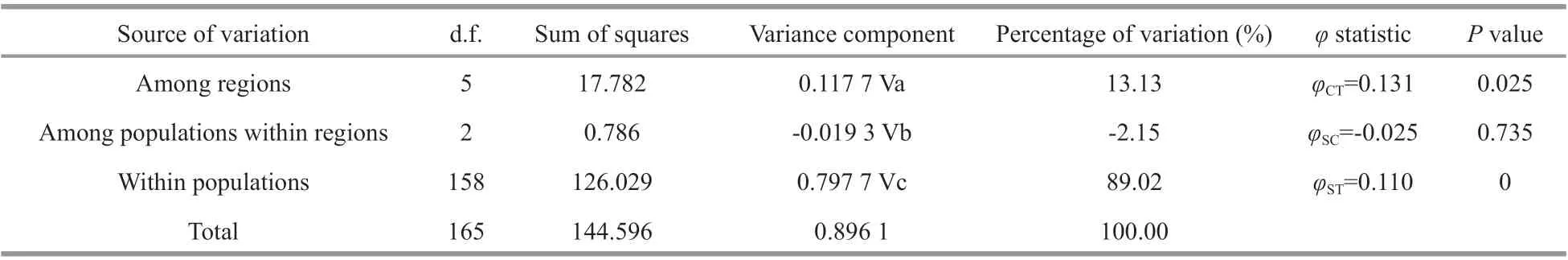
Table 2 Hierarchical analysis of molecular variance (AMOVA) of 16S rRNA haplotypes of A. coerulea
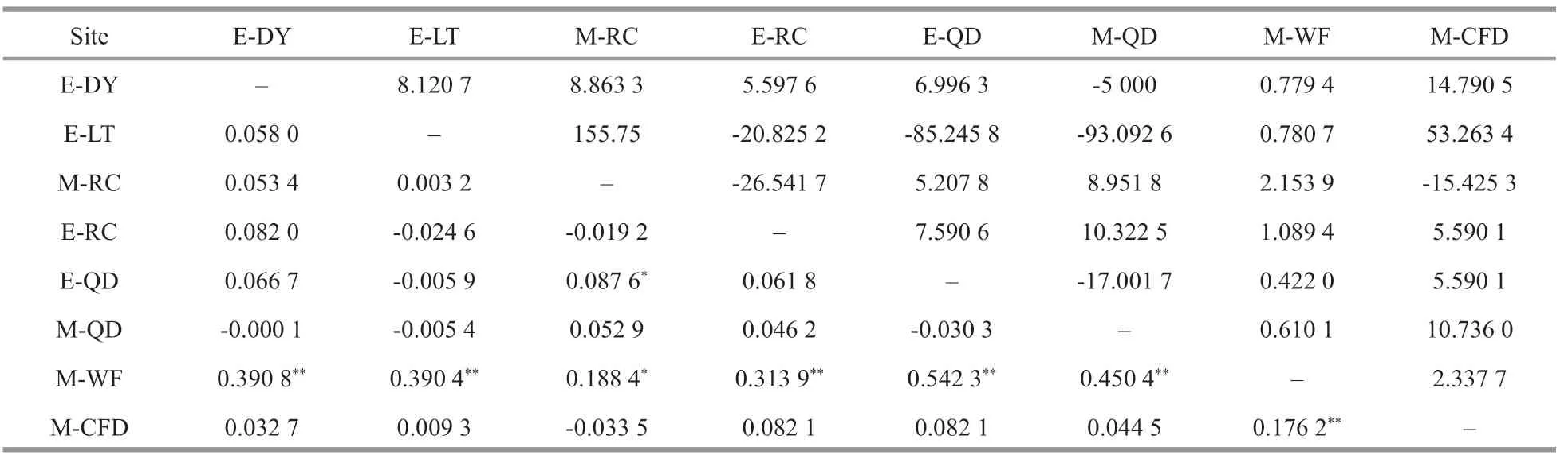
Table 3 Pairwise F st values (below diagonal) and N m values (above diagonal) among populations of A. coerulea based on 16S rRNA
4 DISCUSSION
Most researchers believed that the genusAurelia,which includes 5 valid species, consists of at least 13 species and 16 genetic branches (Dawson and Jacobs,2001; Dawson, 2003; Ki et al., 2008; Scorrano et al.,2017).A.auritahas the widest distribution, except the polar regions; therefore, unidentifiedAureliaspp.have been namedA.auritain most previous reports.Previous research based on 16S rDNA and COI identifiedAureliaspp. in Chinese seas asA.coerulea,but there is no definitive research which determines to which species or genetic branch theAureliaspp.ephyrae found in sea cucumber culture ponds belong.BLASTn analysis indicated that the sequences identified in this study were highly similar (99%) to the sequences ofA.coeruleareported by He et al.(2015) (Genbank accession number: KF962395) and Wang et al. (2013) (Genbank accession number:JX845344), indicating that theAureliaspp. in this study wereA.coerulea.
The genetic diff erentiation betweenA.coeruleaephyrae populations collected in the sea cucumber aquaculture ponds and medusae collected in coastal waters was not significant. Furthermore, among the ephyrae populations in sea cucumber aquaculture ponds and the medusae populations in the coastal waters with close sampling points, the haplotype diversity of ephyrae populations were generally lower than that of medusae populations. The Hd of E-LT was lower than M-CFD, the same results still existed in E-QD and M-QD, E-RC and M-RC. These results suggested that the ephyrae populations in the sea cucumber culture ponds were part of the same genetic group as the medusae populations in the coastal waters. It is likely thatA.coeruleaplanulae of some haplotypes flow in the sea cucumber aquaculture ponds through tides or pumps and settle in the artificial structures because adult medusae ofAureliafrequently occur near theA.japonicusculture ponds during the summer. The water inlets and outlets in the sea cucumber aquaculture ponds are covered with nylon fishing nets to prevent the entrance of potential predators and the escape of farmed sea cucumbers;however, they cannot prevent the exchange ofA.coeruleaplanulae between theA.japonicusculture ponds and coastal waters.
Phylogeographic analysis based on a network relationship diagram suggested that there was no significant correlation between the haplotypes and the distribution of geographic populations. However,pairwise fixation index values showed significant genetic diff erentiation betweenA.coeruleamedusa population of WF and other populations, which means that there was a certain degree of gene flow among the 8 populations, but the dispersal ofA.coeruleaamong WF and other locations was relatively limited. We speculate that this is related to biological characteristics ofA.coeruleaand marine transportation.A.coerulea,as a typical species of zooplankton, its ability to move long distances is relatively weak, and has the characteristics of alternated phenology and weak diff usion ability of polyps in its life cycle. The weak diff usion ability of benthic stages of seasonal meroplanktonic species, such asRhizostomaoctopusandA.coerulea, means they more likely to have distinct genetic population structures than holoplanktonic species, such asPelagianoctiluca(Stopar et al., 2010; Ramšak et al., 2012; Lee et al.,2013). In addition, polyps, the key stage of the expansion of theA.coeruleapopulations, are often attached to artificial structures such as coastal docks and ports. Thus, the areas whereA.coeruleamedusae are gathered in large numbers are usually coastal waters and shoals areas, which are rare in deep-sea areas; this further limits the diff usion range ofA.coeruleapopulations. Similarly, Li et al. (2016)found that there was significant genetic diff erentiation betweenRhopilemaesculentumsampled in the Yellow Sea and Bohai Sea. The Caofeidian population did not show significant genetic diff erentiation from the Yellow Sea populations, which may be due to the developed coastal transportation industry. Bolton and Graham (2006) proposed ballast water and hull carrying in marine transportation play an important role in importing new haplotypes or new species.Compared with Qingdao, Rongcheng and Caofeidian,the scale of coastal transportation in Weifang is still relatively insuffi cient, which allows Weifang population to have a certain degree of gene exchange with other groups, but also limits the frequency and scope of gene flow.
5 CONCLUSION
Based on mitochondrial 16S rRNA region, the ephyrae collected in the aquaculture ponds in the Bohai and Yellow Seas were identified asA.coerulea.The haplotype diversity and nucleotide diversity of the total population ofA.coeruleawere showing a higher haplotype diversity and lower nucleotide diversity. The haplotype diversity of ephyrae populations were generally lower than that of medusae populations with close sampling points. The genetic diff erentiation between ephyrae collected in the sea cucumber aquaculture ponds andA.coeruleamedusae populations collected in coastal waters was not significant. Thus, the ephyrae populations in the sea cucumber culture ponds were part of the same genetic group as the medusae populations in the coastal waters. Phylogeographic analysis of the 16S rRNA region revealed that there was no significant correlation between the haplotypes and the geographic distribution of populations. Pairwise fixation index values showed significant genetic diff erentiation and limited gene flow betweenA.coeruleamedusae population of Weifang and other populations.
6 DATA AVAILABILITY STATEMENT
All data generated and/or analyzed during this study are available from the corresponding author upon request.
 Journal of Oceanology and Limnology2021年3期
Journal of Oceanology and Limnology2021年3期
- Journal of Oceanology and Limnology的其它文章
- Steady increase in water clarity in Jiaozhou Bay in the Yellow Sea from 2000 to 2018: Observations from MODIS*
- Phylogenetic diversity and bioactivity of culturable deepsea-derived fungi from Okinawa Trough*
- Allelopathic eff ects of mixotrophic dinoflagellate Akashiwo sanguinea on co-occurring phytoplankton: the significance of nutritional ecology*
- Investigation of the decline of Ulva prolifera in the Subei Shoal and Qingdao based on physiological changes*
- Effi ciency of phosphorus accumulation by plankton,periphyton developed on submerged artificial substrata and metaphyton: in-situ observation in two shallow ponds*
- Petroleum exploitation enriches the sulfonamide resistance gene sul2 in off shore sediments