
Characterization of microbial communities in sediments of the South Yellow Sea*
2021-06-15 08:08YeCHENSiqiLIXiaoqingXUManmanMATiezhuMIYuZHENZhigangYU
YeCHEN, SiqiLI,Xiaoqing XU , , Manman MA,,, Tiezhu MI , ,,YuZHEN,,,**, Zhigang YU,5
1 College of Marine Life Science, Ocean University of China, Qingdao 266003, China
2 Laboratory for Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology(Qingdao), Qingdao 266237, China
3 Key Laboratory of Marine Environment and Ecology, Ministry of Education, Qingdao 266100, China
4 College of Environmental Science and Engineering, Ocean University of China, Qingdao 266100, China
5 Key Laboratory of Marine Chemistry Theory and Technology, Ministry of Education/ Institute for Advanced Ocean Study,Ocean University of China, Qingdao 266100, China
Abstract Illumina sequencing and quantitative PCR (qPCR) based on the 16S ribosomal RNA (rRNA)gene were conducted to characterize the vertical distribution of bacterial and archaeal communities in the sediments of two sites from the South Yellow Sea. Both bacterial and archaeal communities showed a clear stratified distribution with sediment depth. The microbial communities in the upper layers were distinct from those in the deeper layers; the relative abundances of sequences of Thaumarchaeota,Gammaproteobacteria, and Actinobacteria were higher in the upper than in the deeper sediments, whereas the sequences of Bathyarchaeia, Lokiarchaeota, Euryarchaeota, Chloroflexi, and Deltaproteobacteria were relatively more abundant in the deeper sediments. Sediment depth and total organic carbon (TOC) can significantly influence both the bacterial and archaeal communities. Furthermore, bacterial and archaeal groups potentially involved in nitrogen, sulfur, and methane metabolism were detected in both sites. In our study, both ammonia-oxidizing bacteria ( Nitrospira) and ammonia-oxidizing archaea ( Candidatus Nitrosopumilus) were responsible for ammonia oxidization. Additionally, sulfur-reducing bacteria SEEPSRB1 forming consortia with anaerobic methane-oxidizing archaea ANME-2a-2b were capable of anaerobic methane oxidation (AOM) in the 3400-02 sediment samples.
Key word:microbial community; 16S rRNA gene; high-throughput sequencing; South Yellow Sea; sediment
1 INTRODUCTION
Continental margins are the main interface between terrestrial sediment sources and deep-sea depositional systems (Covault and Fildani, 2014),although they occupy less than 20% of the ocean surface area (Walsh, 1991). Sediment receives microbes and organic matter exporting from the upper water layer, and provides complex nutrients and solid surfaces for the growth of microorganisms(Wang et al., 2012). The biomass and taxon richness of the microbial community in sediments are much higher than those in the corresponding water bodies(Zinger et al., 2011). Marine microbial communities are major engines of biogeochemical processes,including carbon, nitrogen and sulfur, and they probably play pivotal roles in maintaining marine ecosystems to prevent environmental changes such as ocean acidification and global warming (Fuhrman,2009; Graham et al., 2012). Analysis of the microbial community structure in sediments can enhance our understanding of benthic ecosystem processes and the roles that microbes play in overall oceanic processes.
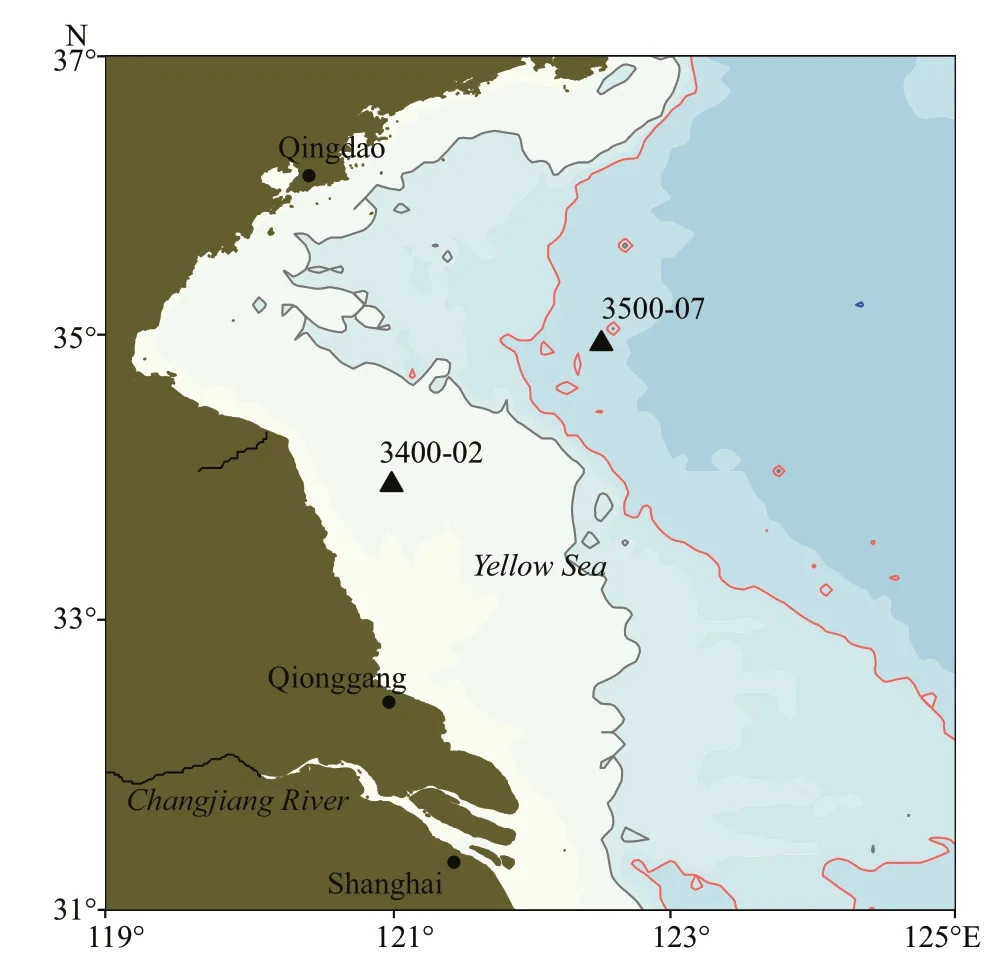
Fig.1 Sampling locations in the South Yellow Sea
The Southern Yellow Sea (SYS) is a semi-enclosed shallow shelf sea surrounded by the Chinese mainland and Korean Peninsula. The sediments of the SYS are mostly terrigenous matter, which is derived mainly from the Chinese mainland and secondarily from the Korean Peninsula (Shi et al., 2003). Simultaneously,the SYS has a complex dynamic system including monsoonal winds, waves, circulation (the Yellow Sea warm current, etc.), and tidal currents (Shi et al.,2003). This complex sedimentary environment in the SYS, including sediment sources and hydrodynamic conditions, can significantly influence the distribution of microbial communities and related biogeochemical processes. However, most studies have primarily focused on the water column and the surface sediments in the SYS (Dong et al., 2014; Liu et al., 2015b), and there is little information on this marine subsurface.Only Qiao et al. (2018) reported the vertical distribution of the bacterial community in the eastern China marginal seas (the SYS and East China Sea(ECS)); however, the potential functional role of archaea in this marine subsurface is still poorly understood. Once thought to inhabit only extreme environments, archaea are now known to inhabit diverse marine environments (DeLong, 1992;Fuhrman and Davis, 1997), where they constitute a relevant fraction of the microbial community (Karner et al., 2001). Archaea members, belonging to Euryarchaeota in marine systems, are recognized to perform the production and anaerobic oxidation of methane (Valentine, 2002; Liu and Whitman, 2008).The oxidation of ammonia to nitrite can be performed by the phylum Thaumarchaeota, which possesses the ammonia monooxygenase subunit A (amoA) genes(Brochier-Armanet et al., 2008; Spang et al., 2010;Berg et al., 2014). Therefore, understanding the variation in archaeal communities in sedimentary environments is crucial for predicting biogeochemical cycles in these environments.
This study was conducted to characterize the structure of the bacterial and archaeal communities at various depths collected in sediment samples from two sites of the SYS. To accomplish this goal, samples were evaluated using 16S rRNA gene high-throughput sequencing and quantitative PCR (qPCR). The present study aims to determine the abundance, diversity, and composition of the microbial community in sediments and their changes with depth, to identify functional microorganisms that can contribute to geochemical cycling in the sediments, and to reveal potential links between the abundance, community composition of bacteria and archaea and environmental factors.
2 MATERIAL AND METHOD
2.1 Sample collection

2.2 Determination of environmental parameters
The environmental parameters (temperature,salinity, and dissolved oxygen concentration) of the bottom water samples were recorded with a CTD(SBE 11 plus, Sea-Bird Electronics, Inc.) in situ.Other parameters were measured in the laboratory.pH was determined using an S479-uMix (METTLER TOLEDO SevenExcellence, Switzerland). The concentrations ofinorganic nitrogen compounds(NH4+, NO3ˉ, and NO2ˉ) were determined by a QuAAtro nutrient autoanalyzer (Seal Analytical Ltd., United Kingdom), and the concentrations of SO42ˉ and Cl–were analyzed using ICS-3000 ion chromatography(Dionex, USA). The elemental concentrations of Fe and Mn were measured by Thermo Scientific iCAP RQ ICP-MS. The content of total organic carbon(TOC) and total nitrogen (TN) in the sediments were determined by elemental analyzer (EURO EA3000,Italy). Methane concentrations were determined using a gas chromatograph (GC; Agilent 7890B) equipped with a Porapark-Q column and a flame ionization detector (FID) by injecting 5 mL of headspace gas.
2.3 DNA extraction and quantitative PCR
Genomic DNA was extracted using the PowerSoil DNA Isolation Kit (Mo Bio, USA) in accordance with the manufacturer’s protocol. The extracted DNA was then stored at -80 °C for further analyses. A SYBR Green method was used to quantify the total abundance of bacterial and archaeal communities,and all qPCR assays were performed on an ABI PRISM®7500 Sequence Detection System (Applied Biosystems, USA). The bacterial 16S rRNA and archaeal 16S rRNA gene copies were quantified using the universal primer pair 338F/806R (Peiff er et al.,2013; Liu et al., 2015a) and U519F/806R (Porat et al.,2010; Shehab et al., 2013), respectively. Each 20-μL mixture contained 10-μL FastStart Universal SYBR Green Master (Rox) (Roche Diagnostics, Germany),0.2-μL bovine serum albumin (BSA), 0.6 μL of each primer and 2.0-μL sediment DNA. Both bacterial and archaeal qPCR amplification conditions were as follows: an initial denaturation step at 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 58 °C for 2 min. All qPCRs were performed in triplicate for each sample. Amplification products were verified by melting curve analysis.
Standard curves for qPCR assays were obtained with serial 10-fold dilutions of plasmids carrying the target gene fragments. Amplification effi ciencies were 90% and 99% for bacteria and archaea, respectively,and a strong linear inverse relation (R2>0.99) was considered credible.
2.4 High-throughput sequencing and data analysis
Five sediment layers (2–4, 8–10, 14–16, 20–22,and 26–28 cm) from site 3400-02 and eight sediment layers (2–4, 8–10, 14–16, 20–22, 26–28, 32–34, 38–40, and 44–46 cm) from site 3500-07 were chosen for bacterial and archaeal 16S rRNA gene tag sequencing. PCR amplifications of the bacterial 16S rRNA gene targeting the V3–V4 region and the archaeal 16S rRNA gene targeting the V4–V5 regions were conducted using primers 338F(5′-ACTCCTACGGGAGGCAGCA) and 806R(5′-GGACTACHVGGGTWTCTAAT) (Lee et al.,2012) and Arch519F (5′-CAGCCGCCGCGGTAA)and Arch915R (5′-GTGCTCCCCCGCCAATTCCT)(Coolen et al., 2004), respectively. The PCR conditions for the 338F/806R primers were as follows: 98 °C for 5 min; 24 cycles of 98 °C for 30 s,52 °C for 30 s and 72 °C for 45 s; and then 72 °C for 5 min. The PCR conditions for the Arch519F/Arch915R primers consisted of 98 °C for 5 min; 35 cycles of 98 °C for 30 s, 65 °C for 30 s and 72 °C for 45 s; followed by 72 °C for 5 min. PCR was carried out on an ABI 2720 (Applied Biosystems, USA),and PCR products were electrophoresed on a 1.2%polyacrylamide gel. The target fragment was recovered with Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad,CA, USA). High-throughput sequencing of the 16S rRNA genes were conducted on the Illumina MiSeq platform (Personal Biotechnology Co., Ltd.,Shanghai, China).
Sequenced reads were de-multiplexed, qualityfiltered, and processed using the QIIME software package (http://qiime.org/) according to the following three criteria (Caporaso et al., 2012). The paired-end reads were joined with at least a 10-bp overlap and no mismatches using FLASH (Magoč and Salzberg,2011). Sequences were clustered into operational taxonomic units (OTUs) based on their sequence similarity at a 97% dissimilarity level using UCLUST.A representative sequence for each OTU was assigned against the SILVA SSU Ref NR database (https://www.arb-silva.de/no_cache/download/archive/release_132ARB_files/) via QIIME.
2.5 Statistical analyses
Any OTU containing less than 0.001% of the total sequences in samples was discarded to reduce the eff ect of spurious OTUs on further analysis (Bokulich et al., 2013). To fairly compare all the samples at the same sequencing depth, sequences were randomly reduced based on the smallest read numbers in each sample. Then, the diversity indices, including Good’s coverage, Shannon, Simpson, and Chao 1, were calculated using the QIIME software package. For beta diversity, the Bray-Curtis dissimilarity-based dendrogram was plotted to examine the diff erences in bacterial and archaeal communities between the sediment samples. Comparisons of parameters between the two sites were performed using SPSS by one-way ANOVA. The Spearman correlation test was used to evaluate correlations between microbial abundance, diversity, and the 20 most abundant bacterial and archaeal genera with environmental parameters. Then, heatmaps were constructed to visualize Spearman’s correlation using an online tool(http://www.lc-bio.cn).
3 RESULT
3.1 Environmental parameter characterization

3.2 Abundance of total bacterial and archaeal 16S rRNA genes
Quantitative PCR was used to detect the abundance of the bacterial and archaeal communities based on the 16S rRNA genes (Fig.2). The observed bacterial 16S rRNA gene abundance at 3400-02 and 3500-07 ranged from 8.50×107to 3.71×108copies/g and 1.25×108to 1.03×109copies/g (wet weight),respectively (Fig.2a). The bacterial abundance showed a decreasing trend with sediment depth at both sites. The relative abundance of bacteria in the total microbial community ranged from 97.12% to 99.41% at site 3400-02 and 93.00% to 99.78% at site 3500-07, and the relative bacterial abundance at site 3500-07 decreased with sediment depth (Fig.2b).
The observed archaeal 16S rRNA gene abundance ranged from 5.38×105to 7.93×106copies/g and 1.55×106to 2.92×107copies/g at 3400-02 and 3500-07 (wet weight), respectively (Fig.2a). In contrast to the vertical variation of bacteria abundance along the sediment depth, the archaea abundance decreased with the sediment depth in the upper 10 cm at site 3400-02 and then showed marked fluctuations. At site 3500-07 in contrast, the archaea abundance increased with sediment depth. For the two sites, the relative abundance of archaea within the total microbial community was 0.59%–2.88% at site 3400-02 and 0.22%–7.00% at site 3500-07, and the relative archaeal abundance at site 3500-07 increased with sediment depth (Fig.2b).
3.3 Bacterial and archaeal diversity and richness
A total of 419 741 high-quality bacterial 16S sequences and 510 751 archaeal 16S sequences were recovered from Illumina-based analysis (Tables 1 &2). The number of OTUs varied from 1 809 to 2 696 per sample and 804 to 3 005 per sample at a 97%sequence similarity level for the bacterial and archaeal communities, respectively. Good’s coverage of bacterial and archaeal communities at site 3400-02 and 3500-07 ranged from 93.21%–100.00% and 95.70%–100.00%, respectively, which indicated that the sequencing depth was suffi cient to cover most of bacterial and archaeal communities. At the corresponding sediment layer of the two sites, the bacterial and archaeal diversity (Shannon-Wiener and Simpson’s diversity index) was higher at site 3500-07 than 3400-02, with the exception of the bacterial community in the 14–16-cm layer. At site 3400-02,the bacterial Shannon-Wiener index values were higher than the archaeal community in the corresponding sediment layer, while at site 3500-07,the bacterial Shannon-Wiener index values were higher than the archaeal community in the 2–22-cm layers but became lower in the deeper sediments (26–46 cm).With respect to bacterial diversity, Simpson’s diversity index was negatively correlated with sediment depth (P<0.05). With respect to archaeal diversity, both Simpson’s diversity index and Shannon-Wiener index were positively correlated with sediment depth, TN, and SO42ˉ (P<0.05) but negatively correlated with NO3ˉ (P<0.05), and the archaeal Chao 1 index was positively correlated with sediment depth and TN (P<0.05) but negatively correlated with NO3ˉ (P<0.05) (Fig.3).
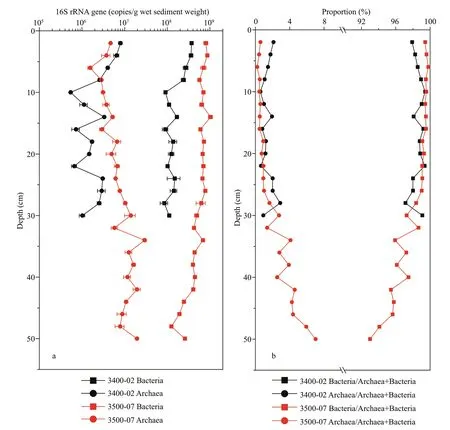
Fig.2 Vertical profiles of total bacterial and archaeal 16S rRNA gene copy numbers in sediment cores at the two sites (a) and the proportion of bacterial abundance or archaeal abundance relative to the total microbial abundance in sediment cores at the two sites (b)
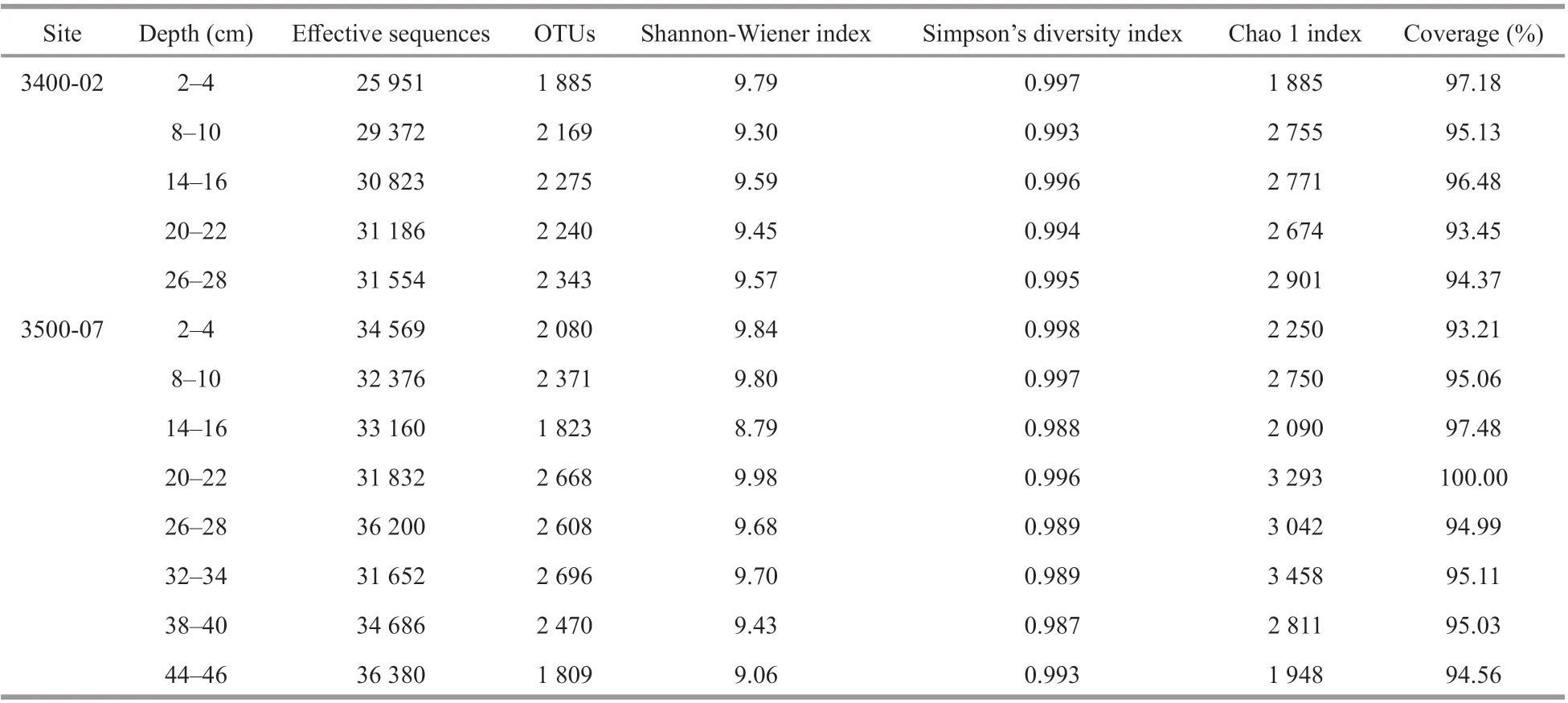
Table 1 Vertical distribution of sediment bacterial richness and diversity estimates based on the 16S rRNA gene
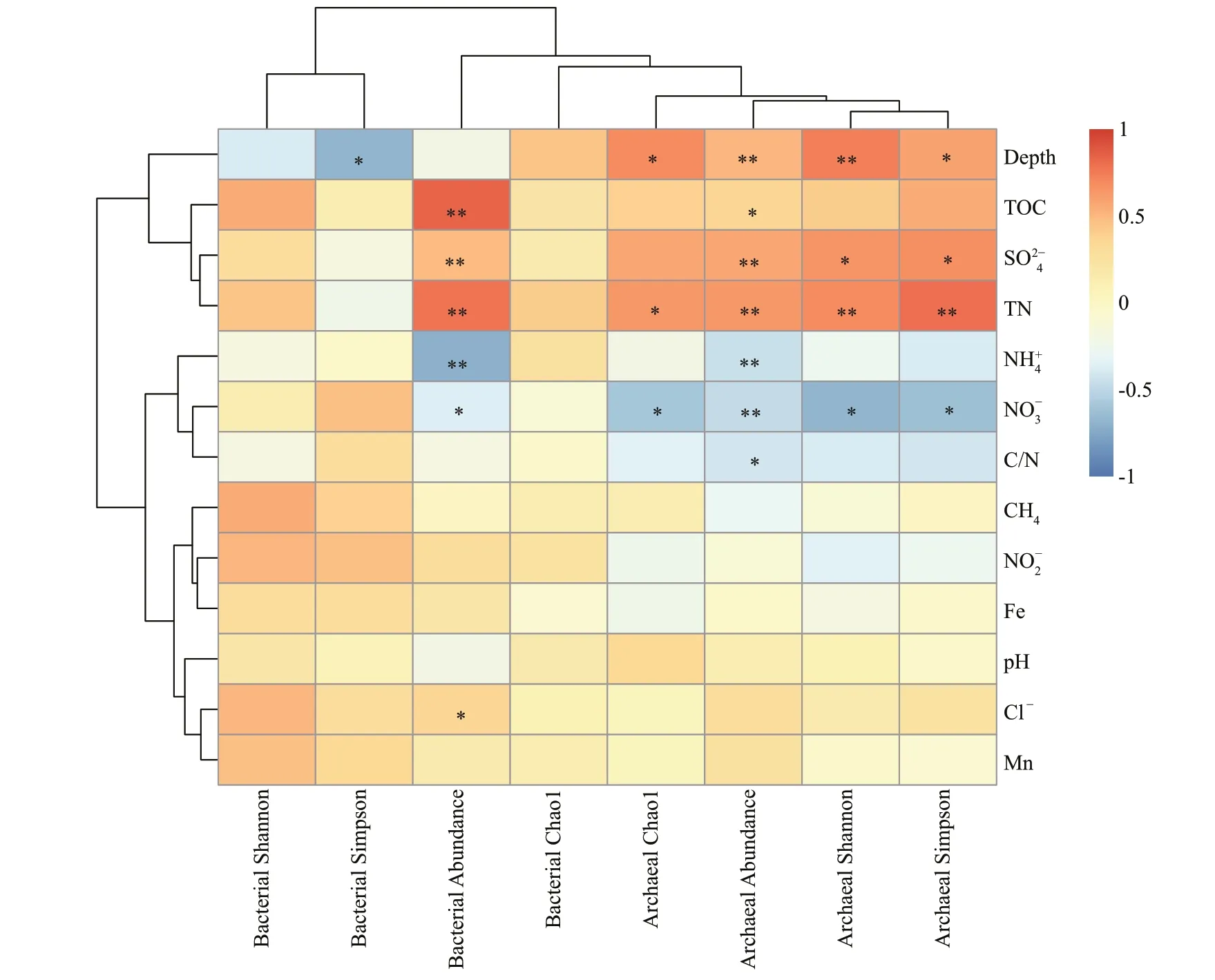
Fig.3 Heatmap of Spearman’s correlation coeffi cients between the abundance and diversity of bacterial and archaeal communities and environmental factors
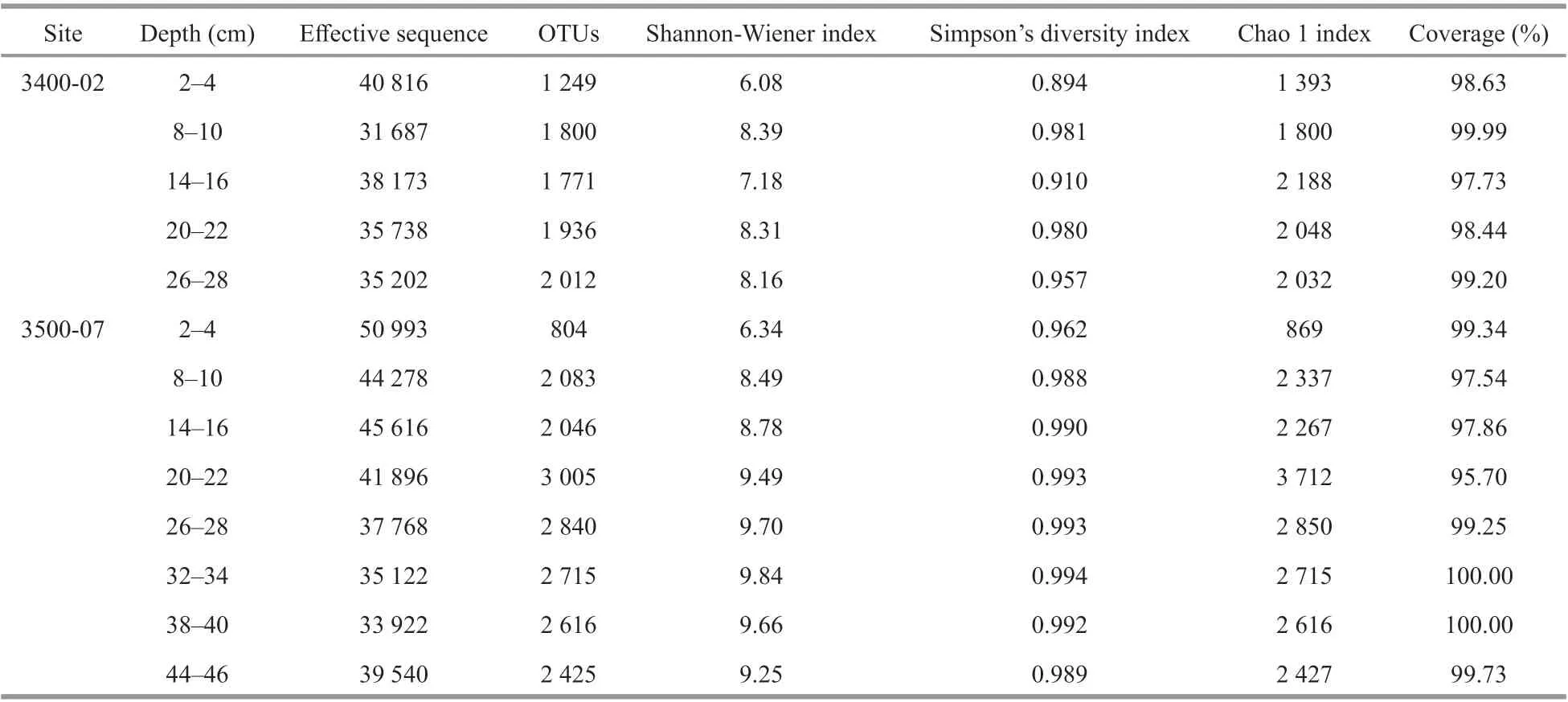
Table 2 Vertical distribution of sediment archaeal richness and diversity estimates based on the 16S rRNA gene
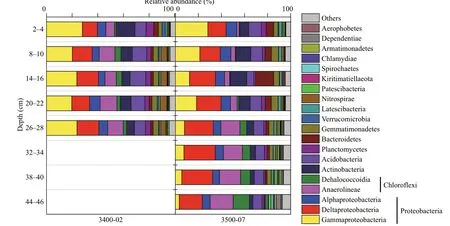
Fig.4 Relative abundance of the bacterial community (including classes of Proteobacteria and Chloroflexi) observed in sediment cores collected from the SYS at the phylum level with an abundance >1%
3.4 Bacterial 16S rDNA analysis
The composition of bacterial communities of sediment samples from the SYS is shown in Fig.4.Most of the bacterial sequence reads were associated with Proteobacteria, accounting for 43.41% of all the sequences, followed by Chloroflexi (16.35%),Acidobacteria (9.31%) and Actinobacteria (8.18%).Among the Proteobacteria, Deltaproteobacteria(19.33%) and Gammaproteobacteria (16.94%) were the most abundant, followed by Alphaproteobacteria(7.14%). Chloroflexi mainly consisted of two classes: Anaerolineae (10.84%) and Dehalococcoidia(4.10%).
At site 3400-02, Gammaproteobacteria represented the predominant group in each sediment layer,followed by Deltaproteobacteria and Anaerolineae.At site 3500-07, sequences from Gammaproteobacteria dominated in the 2–10-cm sections and decreased with sediment depth. The relative abundances of Deltaproteobacteria increased with sediment depth and dominated in the 14–46-cm sections. Additionally,Anaerolineae and Dehalococcoidia belonging to the Chloroflexi were enriched in the deeper sediments(26–46 cm) of site 3500-07.
3.5 Archaeal 16S rDNA analysis
Nine diff erent archaeal phyla were identified in the sediment samples (Fig.5a). Crenarchaeota (44.12%)and Thaumarchaeota (32.58%) were the most abundant phyla of archaeal sequences, followed by Asgardaeota (11.97%) and Euryarchaeota (5.67%).Within the Crenarchaeota, Bathyarchaeia (43.81%)(Formerly named MCG) had the highest relative abundance (Fig.5b). Sequences belonging to Bathyarchaeia had lower abundances in the surface sediments of sites 3400-02 (18.93%) and 3500-07(3.64%); however, they predominated in the subsurface sediment layers of the two sites, ranging from 36.58% to 60.83% (3400-02) and 36.81% to 57.28% (3500-07). Nitrososphaeria was the most abundant class within the Thaumarchaeota, which was found to predominate in the surface sediment of the two sites and to decrease with sediment depth at site 3500-07. Lokiarchaeia (11.48%) was the most dominant class within Asgardaeota, and it was mainly present in the subsurface sediments (8–46 cm) of site 3500-07. Among Euryarchaeota, the majority of sequences were assigned to Thermoplasmata, which was particularly enriched in deeper sediment layers(26–46 cm) at site 3500-07 (Fig.5b).
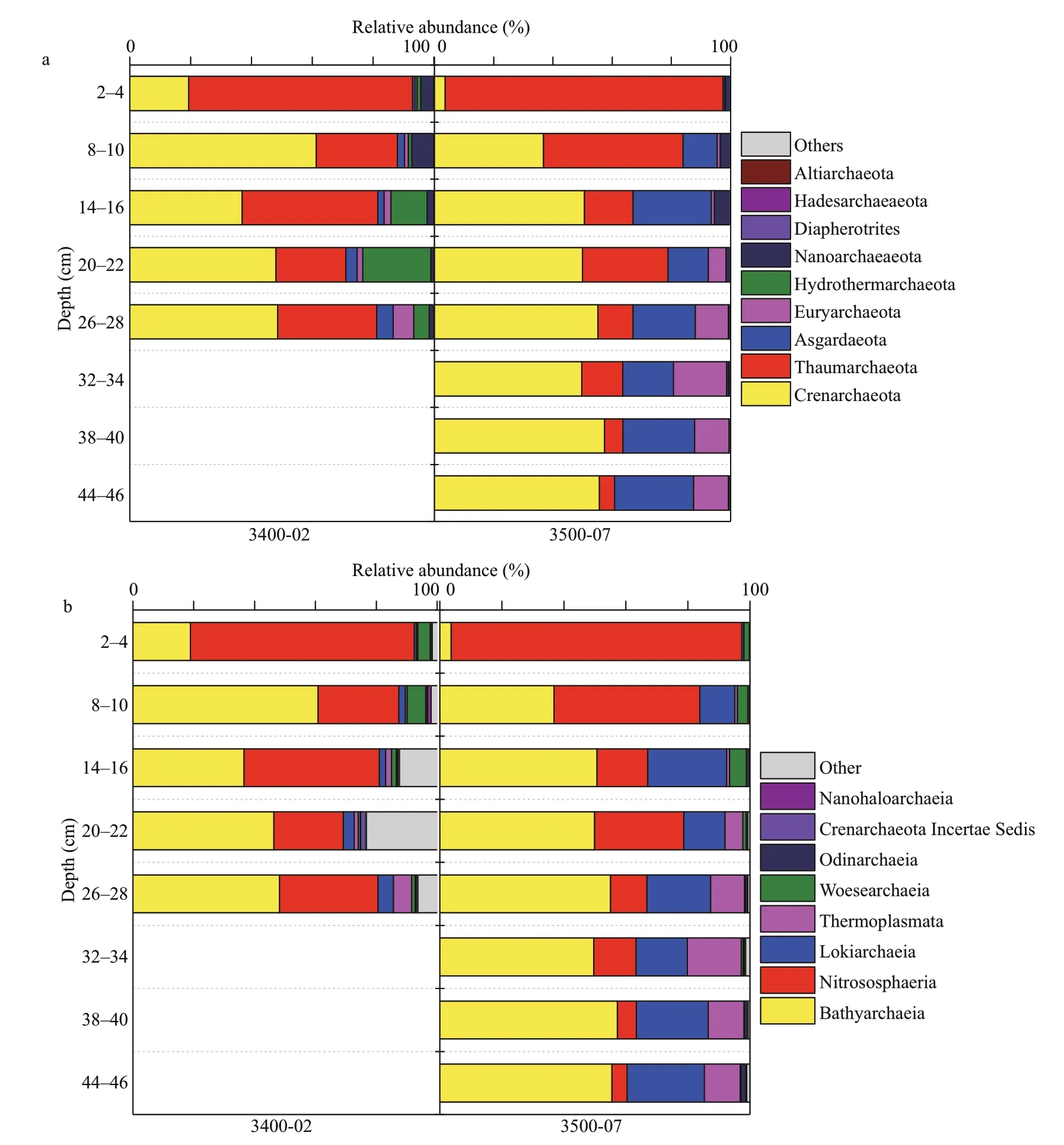
Fig.5 Relative abundance of archaeal groups observed in sediment cores collected from the SYS
3.6 Community comparison and relationships between community structure and environmental factors
The bacterial and archaeal community structures clearly diff ered between the two sites and among diff erent sediment depths, as displayed by the clustering analysis (Fig.6). Overall, the bacterial and archaeal community structures in the sediment layers from site 3400-02 formed distinct clusters, and those in the sediment layers from site 3500-07 tended to cluster together. The upper layer samples (2–22 cm)of site 3500-07 clustered, whereas the deep layer samples (26–46 cm) tended to cluster together for the bacterial community. For the archaeal community, the surface sediment layer of site 3500-07 was obviously diff erent from the other layers, and the other layers clustered into two groups: the upper layer samples(8–22 cm) clustered into one group, whereas the deep layer samples (26–46 cm) clustered together.

Fig.6 UPGMA hierarchical clustering based on a Bray-Curtis distance matrix for the bacterial composition (a) and archaeal composition (b) at the OTU level
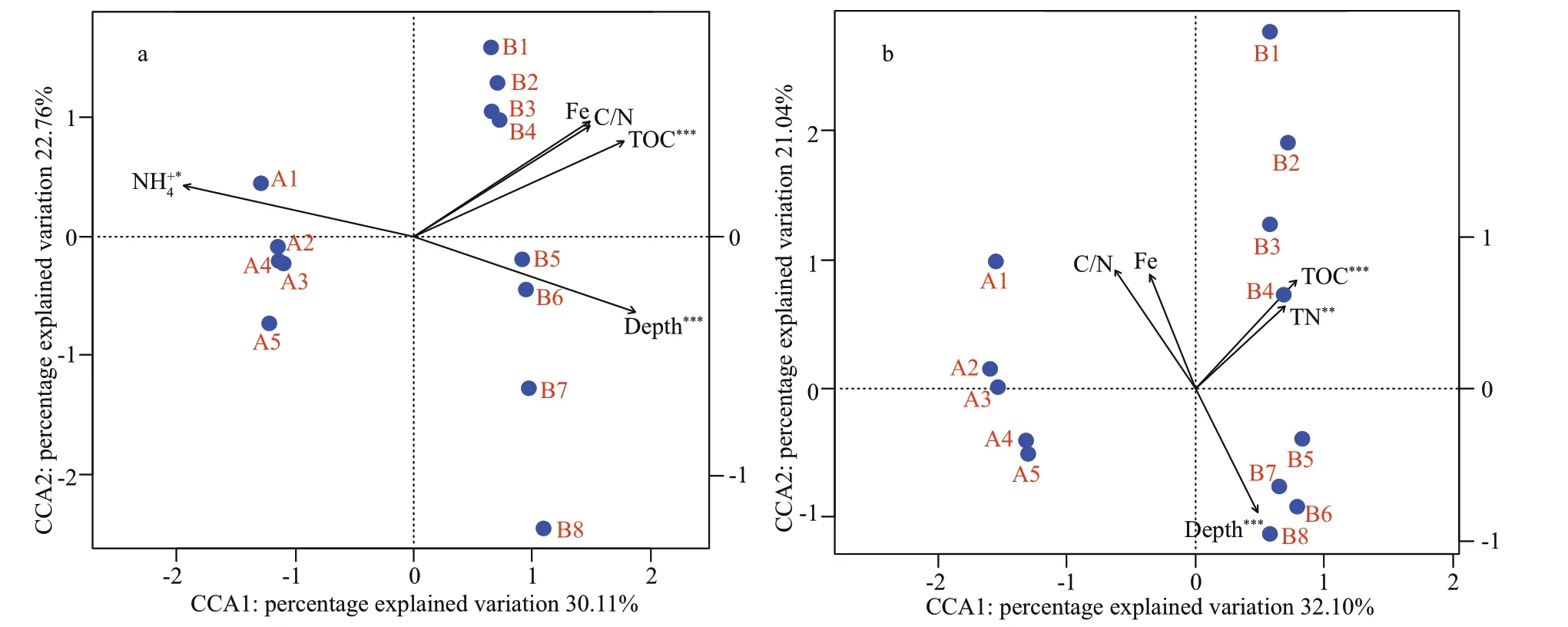
Fig.7 Canonical correspondence analysis (CCA) biplot of environmental parameters and microbial community structure at the OTU level
Biota-environment (BIOENV) identified that sediment depth, TOC, C/N, NH4+, and Fe were most strongly correlated with the bacterial community,while sediment depth, TOC, TN, C/N, and Fe were most strongly correlated with the archaeal community. These variables explained 52.87% and 53.14% of the variation in the bacterial and archaeal communities, respectively, as determined by CCA(Fig.7). The composition of the bacterial community was significantly correlated with sediment depth,TOC and NH4+and the archaeal community was significantly correlated with sediment depth, TOC and TN via Monte Carlo permutation significance tests.
3.7 Microbes involved in biogeochemical cycles
The 16S rRNA genes showed that ammoniaoxidizing bacteriaNitrosospiraand ammoniaoxidizing archaeaCandidatusNitrosopumiluswere the dominant groups associated with ammonia oxidation at the two sites. The relative abundances ofNitrosospira(ammonia-oxidizing bacteria (AOB))andCandidatusNitrosopumilus(ammonia-oxidizing archaea (AOA)) were higher in the upper sediment layers at both sites, and their proportions decreased with increasing depth (Fig.8a & b). Furthermore, the proportion of AOA in the archaeal community was much higher than AOB in the bacterial community at each site. The bacterial genusNitrospirawas the main group associated with nitrate oxidation and exhibited relative abundances of 0.14%–0.29% and 0.02%–1.17% at 3400-02 and 3500-07, respectively (Fig.8c).With respect to denitrifying bacteria,Bacilluswas the main group in most sediment layers, except the 2–4 cm and 20–22 cm at site 3400-02, in whichVibrioorMicrobacteriumwas the predominant group (Fig.8d).
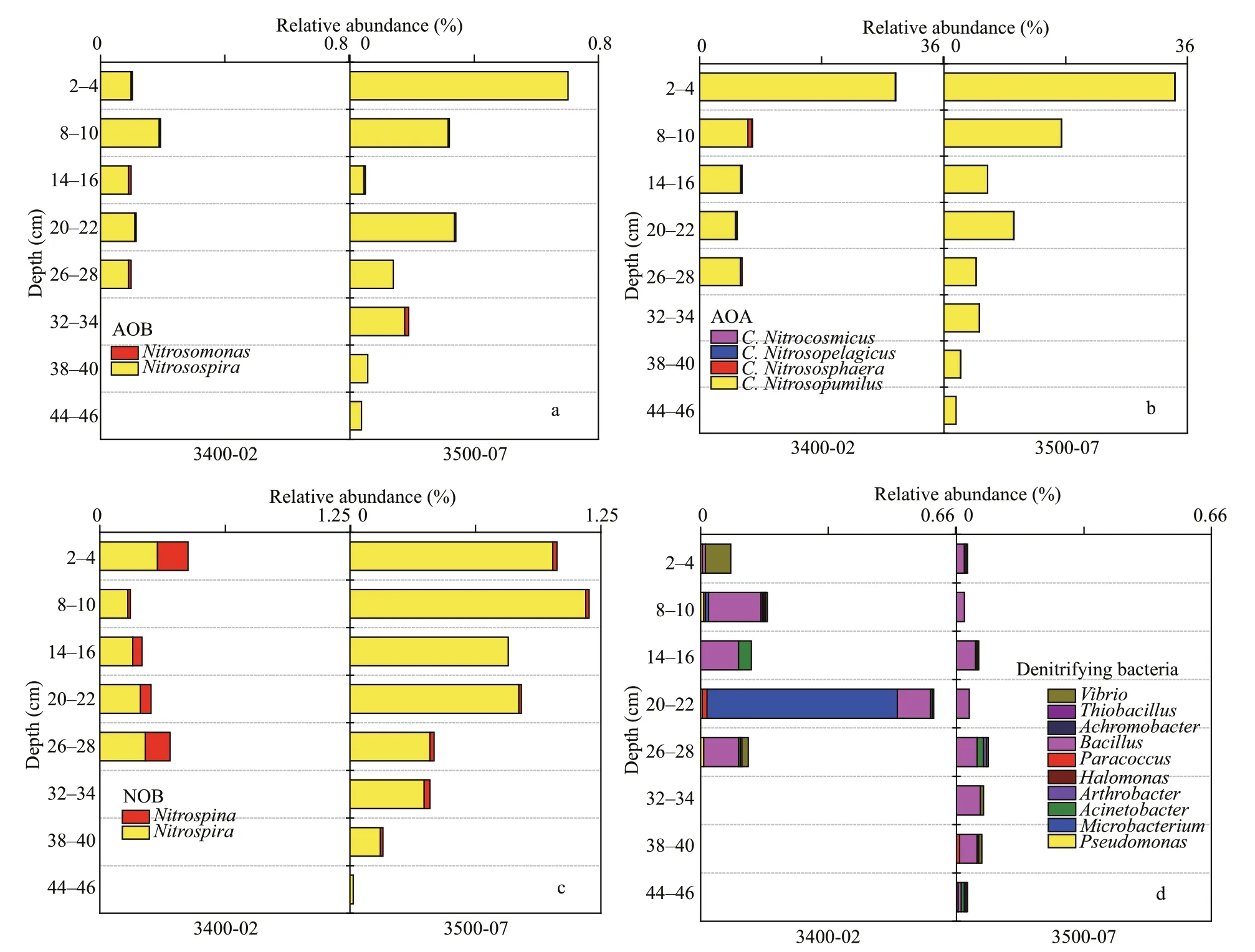
Fig.8 Relative abundances of microbes involved in the nitrogen process at the two sites
Thirty-four genera (Desulfatiglans,SEEP-SRB1,Desulfuromonas, etc.) of sulfate-reducing bacteria(SRBs) affi liated with Delta-proteobacteria and one genus (Thermodesulfovibrio) belonging to Nitrospirae were detected to participate in sulfate reduction.DesulfatiglansandSEEP-SRB1dominated at the two sites, and their relative abundance increased with sediment depth (Fig.9a). Sulfur-oxidizing bacteria(SOB) were affi liated with Alpha-, Delta-proteobacteria,Epsilon-proteobacteria, and Spirochaetae (Fig.9b).The genusSpirochaetawithin Spirochaetae represented the dominant SOB at site 3400-02 and deeper sediment layers (26–46 cm) at site 3500-07 and tended to increase with sediment depth at each site. The SOB bacteriaAqS1were dominant in the upper sediment layers (2–22 cm) of site 3500-07.
The methanogens had a higher relative abundance at site 3400-02, accounting for 0.15%–0.66% (0.35%on average), than at site 3500-07, with a relative abundance of 0.01%–0.17% (on average 0.05%)(Fig.10a). The composition of methanogens varied significantly between the two sites. Fifteen genera(Methanosarcina,Methanolobus,Methanobacterium,etc.) were discovered at site 3400-02, whereas seven genera (Methanococcoides,Methanosarcina,Methanobrevibacter, etc.) were discovered at site 3500-07. Among the methanogens,Methanosarcina,with a relative abundance of 0.13% (on average), was the dominant methanogen at site 3400-02, whereasMethanococcoides, with a relative abundance of 0.04% (on average), was the dominant methanogen at site 3500-07, except at 14–16 cm, in whichMethanosarcinawas the dominant group. Sequence analysis of the 16S rRNA genes also detected a minor fraction of microbial communities involved in methane aerobic oxidation.Methylobacteriumwas the predominant methanotroph at site 3500-07, while the methanotrophs at site 3400-02 consisted of three genera:Methylobacterium,Methylomarinum, andMethyloprofundus(Fig.10b). Anaerobic methane-
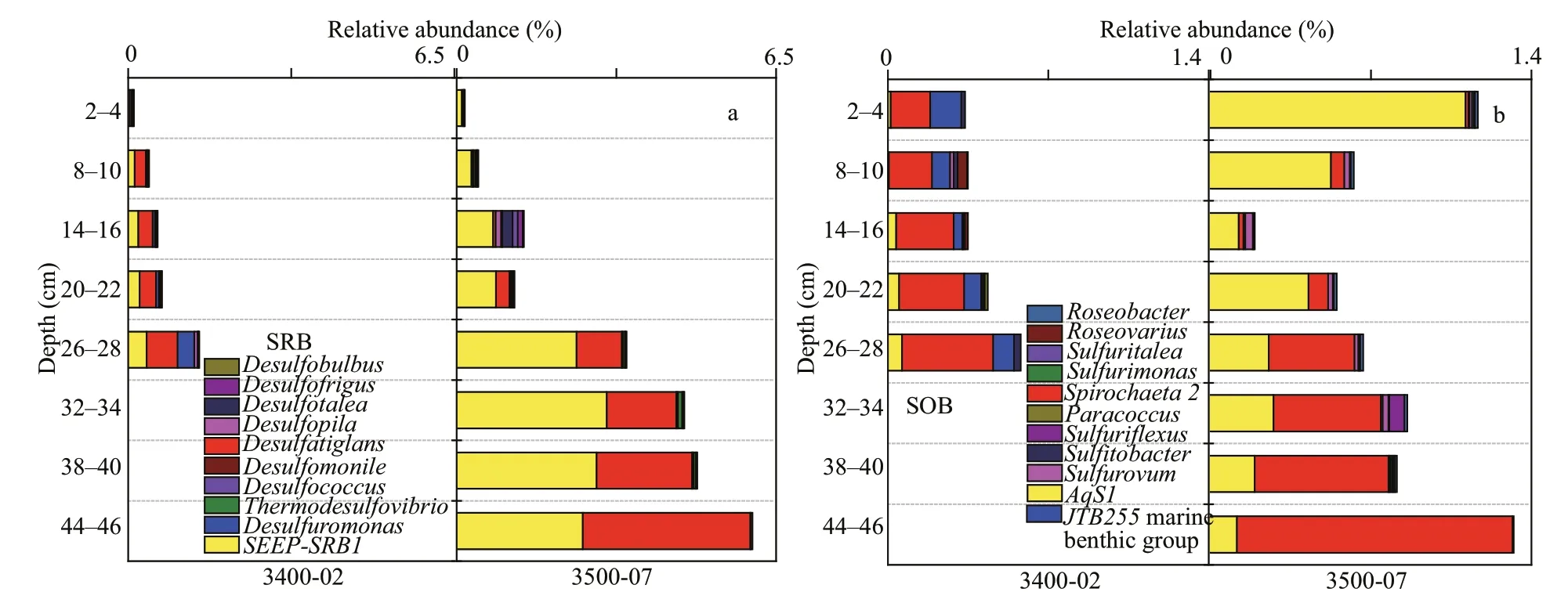
Fig.9 Relative abundances of microbes involved in the sulfur process
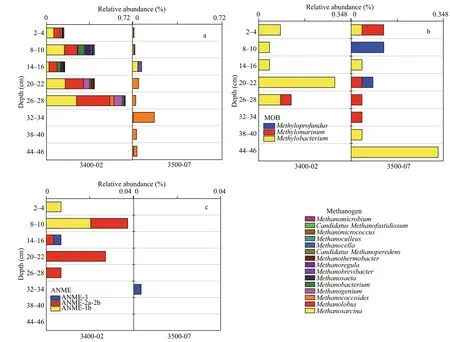
Fig.10 Relative abundances of microbes involved in the methane process at the two sites
a. sulfur-reducing bacteria (SRB); b. sulfur-oxidizing bacteria (SOB).oxidizing archaeas (ANMEs), including ANME-1b,ANME-2a-2b, and ANME-3, associated with CH4anaerobic oxidation, were detected in the sediment samples of site 3400-02 (Fig.10c). ANME-1b dominated in the 2–4-cm and 8–10-cm layers, while ANME-2a-2b dominated in the other sediment layers.At site 3500-07, only a minor fraction of ANME-3 was detected in the 32–34-cm layer.
4 DISCUSSION
4.1 Bacterial and archaeal abundance in sediments of the SYS
In this study, the composition and distribution of bacterial and archaeal communities were characterized in the sediments of two sites of the SYS. The abundance of the bacterial community was significantly higher than the abundance of the archaeal community at each site (one-way ANOVA,P<0.01)(Fig.2a). This result is in agreement with many previous studies, which demonstrated that bacteria rather than archaea dominate in sediments from various marine environments (Schippers and Neretin,2006; Schippers et al., 2012; Liu et al., 2015b).Organic matter availability in marine sediments is recognized as an important factor that aff ects the abundance of the microbial community (Säwström et al., 2016; Qiao et al., 2018). In this study, the TOC and TN contents at site 3500-07 were 2.35–7.73 times and 1.05–14.75 times higher than at site 3400-02 in the corresponding layer. Accordingly, the bacterial and archaeal 16S rRNA gene abundances at site 3500-07 were 1.18–6.80 times and 0.10–12.65 times higher than at site 3400-02 in the corresponding layer, with the exception of the archaeal community in the upper 8-cm layers. It was indicated that fresh and marine organic matter (higher TOC and TN) can support higher microbial abundance.
4.2 Bacterial and archaeal community compositions in sediments of the SYS
The bacterial community composition at each site was dominated by Proteobacteria, which mainly consisted of the classes Alpha-, Delta- and Gammaproteobacteria (Fig.4). The phylum Proteobacteria, as the most dominant group, was also observed in shallow sea and deep-sea sediments (Zhu et al., 2013; Liu et al., 2015b). Most members of Delta-proteobacteria are well known as SRBs (Varon-Lopez et al., 2014), while groups within the Gammaand Alpha-proteobacteria have been shown to perform sulfur oxidation (Bagchi et al., 2005; Lenk et al.,2011). Previous studies have demonstrated their dominance in the sediments of the northern Chinese marginal seas, implying that sulfate and/or sulfurrelated processes could be the most important biogeochemical reactions in these areas (Zhu et al.,2013; Liu et al., 2015b; Zhang et al., 2017; Qiao et al.,2018). Consistent with previous studies of marine sediments (Lentini et al., 2014; Mahmoudi et al.,2015; Qiao et al., 2018), Chloroflexi dominated in deep sediments (3500-07: 26–46 cm). Compared with other microorganisms, Chloroflexi can better utilize recalcitrant organic matter buried in deep sediments,which likely explains their higher abundance in such niches (Wilms et al., 2006; Zhou et al., 2017). In this study, the major component of Chloroflexi was Anaerolineae, which contains members of gramnegative chemoorganotrophic bacteria and can only grow under strict anaerobic conditions as previously described (Yamada et al., 2006). Interestingly, the phylum Chlamydiae accounted for 0.11%–0.22% and 0.08%–1.23% of the total bacterial sequences at site 3400-02 and 3500-07, respectively (Fig.4).Traditionally, Chlamydiae are regarded as important pathogens and symbionts with eukaryotic hosts.However, a recent report showed that in Arctic Mid-Ocean, Chlamydiae dominated in deep anoxic marine,with relative abundances up to 43% of the bacterial community (Dharamshi et al., 2020). The features of host dependency in marine-sediment Chlamydiae suggested that these members play an important and overlooked ecological role in marine ecology and may have alternate lifestyle strategies (Dharamshi et al., 2020).
In our study, the archaeal community showed a clear stratified distribution as the aerobic group.Thaumarchaeota was abundant in the surface layer and decreased rapidly with depth, whereas Bathyarchaeia and Lokiarchaeia tended to be dominant in the lower layers and increased accordingly along the depth profiles (Fig.5b). Sequences affi liated with Thaumarcheaota accounted for a high percentage of the total archaeal sequences in the surface sediments of the two sites. Thaumarchaeota is the most abundant archaea on earth and widely distributed in freshwater,soils, marine waters, and sediments (Off re et al.,2013). Species within the Nitrososphaeria class of Thaumarchaeota have been identified as aerobic ammonia-oxidizers (Stahl and de la Torre, 2012). The high relative abundance of Nitrososphaeria observed herein suggested that this group could play a major role in nitrification and nitrogen cycling in sediments.Bathyarchaeia (formerly named MCG) and Lokiarchaeia (formerly named DSAG or MBGB)have been proposed as major groups in heterotrophic metabolism of organic matter in anoxic sediments(Inagaki et al., 2006; Nunoura et al., 2012). Moreover,previous studies have shown that members of the Bathyarchaeia are anaerobic methanotrophs (Evans et al., 2015), methanogens (Evans et al., 2015),acetogens (He et al., 2016b; Lazar et al., 2016) or anaerobic heterotrophs (Lloyd et al., 2013, Lazar et al., 2016), indicating that Bathyarchaeia can play an important role in the organic carbon cycle.Lokiarchaeia or MBGB has been suggested to play a role in biogeochemical processes such as sulfate reduction and methane oxidation (Inagaki et al.,2006). The majority of sequences of Euryarchaeota are affi liated with Thermoplasmatales, which are regularly detected in marine sediments (Durbin and Teske, 2012). Microorganisms from the archaeal class Thermoplasmata have been found to be linked to methanogenic activities (Poulsen et al., 2013). Taken together, our results indicate that the complex archaeal communities in our given sediments might be involved in diverse metabolic processes, especially carbon and nitrogen cycling.
4.3 Archaea plays important roles in deep-water sediments
In our study, the bacterial community abundance and their ratio within the total microbial community showed a slightly decreasing tendency from the surface layers down to the subsurface layers of each site (Fig.2).With respect to the archaeal community abundance, a diff erent distribution pattern was identified at site 3500-07. The abundance of archaea and their ratio within the total microbial community increased with sediment depth (Fig.2). The changing pattern for bacterial and archaeal abundance observed herein was consistent with many previous reports in the shallow sediments of marine benthic habitats (Urakawa et al., 2000; Jiang et al., 2011; Zhang et al., 2017; Pala et al., 2018; Qiao et al., 2018). Heterotrophic bacteria respiration consumes oxygen rapidly in upper layers of the sediment (Qiao et al., 2018). As a result, the microbial carbon oxidation rates declined in deeper sediments followed by a subsequent decrease in bacterial abundance (Qiao et al., 2018). However, archaea can unite by a universal ecological ability to adapt to energy stress in the subsurface environment where food and energy resources are poor (Gold, 1992). Moreover, the archaeal community had a higher richness and diversity in the deeper sediments (3500-07: 20–46 cm). Archaea Bathyarchaeia, Lokiarchaeia, and Thermoplasmata were enriched in these deeper water sediments, leading to a higher archaeal diversity. These members belong to anaerobic heterotrophs that can consume buried carbon(Biddle et al., 2006; Lin et al., 2015), indicating that they could contribute most to the anaerobic degradation of organic matter in such niches.
4.4 Microbes with potential ecological functions
Although accurate measures of metabolic functions are impossible based on 16S rRNA gene sequencing data alone (Tringe and Hugenholtz, 2008; Sun et al.,2018), we can use 16s rRNA taxonomies to predict the functional potential of microbes. Ecological function can be inferred from a genus level phylogeny,which provides better phylogenetic resolution than higher taxonomic categories (Ho et al., 2016; Sun et al., 2018). Nitrification is a two-step biological process that converts ammonia to nitrite and subsequently to nitrate. Ammonia oxidation is generally regarded as the rate-limiting step in nitrification and is carried out by both AOB and AOA.In this study, 16S rRNA gene sequencing analysis of microbial communities indicated that the majority of AOB fell within theNitrosospiraand that the majority of AOA fell within theCandidatusNitrosopumilusat both sites (Fig.8a & b). This result is consistent with findings showing that AOBNitrosospiraand AOANitrosopumilususually dominate the ammoniaoxidizing community in marine systems (He et al.,2016a, 2018; Yu et al., 2016).
Sulfate reduction, mediated by SRB groups, is the dominant organic matter mineralization process in sulfate-rich shelf sediments. In this study, the generaDesulfatiglansandSEEP-SRB1dominated at both sites (Fig.9a).SEEP-SRB1forming consortia with methane-oxidizing archaea ANME-2a-2b are capable of anaerobic methane oxidation (AOM) by using sulfate as the terminal electron acceptor (Timmers et al., 2016). ANME-2a-2b was also detected in the samples of site 3400-02 except for the surface layer(0–2 cm), suggesting that sulfate-dependent anaerobic methane oxidation (SAMO) might occur at this site.Robador et al. (2016) found thatDesulfatiglanswas one of the major groups of sulfate-reducing bacteria in polar, temperate, and tropical marine sediments.Groups withinDesulfatiglanscan completely oxidize organic matter such as phenol, benzoate, or aromatic compounds to carbon dioxide (Suzuki et al., 2014). In our study,AqS1(of the Gammaproteobacteria class)andSpirochaeta 2(of the Spirochaetae phylum) were the dominant sulfur-oxidizing bacteria. Previous studies have indicated thatAqS1can also take part in carbon monoxide oxidation and inorganic phosphate assimilation (Gauthier et al., 2016). Interestingly, the trends of the relative abundance ofSpirochaeta 2at the two sites were consistent with those of the relative abundance of SRB. This finding can be explained by the possible coexistence of Spirochaeta phylotypes with SRB by supplying them with carbon sources and electron donors (acetate, H2, and CO2) (Berlanga et al., 2008).
Methanogenesis is the final degradation process of organic matter in anoxic environments. SRB usually outcompete methanogens for substrates (H2, CO2,and acetate) due to their higher energy yield and higher substrate affi nity in sulfate-rich environments(Schönheit et al., 1982; Lovley and Klug, 1983). In our study, a low abundance of methanogen groups was detected due to a high level of sulfate.Methanosarcinawas predominant at site 3400-02(Fig.10a), which is made up of highly versatile species that are capable of methanogenesis using various substrates (acetate, C1compounds, CO2/H2) (Liu and Whitman, 2008). The genusMethanococcoidespredominated at site 3500-07 (Fig.10a) and can only use C1compounds to produce methane (Liu and Whitman, 2008). The dominance of methylotrophic methanogens in our study implies that methylotrophic methanogenesis might be the main methanogenic pathway at site 3500-07. Studies have shown that methylotrophic methanogenesis dominated CH4production in shallow marine sediments with higher levels of sulfate in Aarhus Bay, Orca Basin, the Gulf of Mexico, and the western Mediterranean Sea (Xiao et al., 2018; Zhuang et al., 2018), because methylotrophic methanogens can be competitive when they compete with sulfate reducers for methylated substrates (e.g., methanol, methylamines or other C1 compounds). However, according to the correlation analysis, there was no significant correlation between the relative abundance of methanogens and the concentration of CH4in our studied sediments (R=-0.223,P>0.05), probably because the CH4concentration was mediated by both methanogen and methanotrophs. Both aerobic methanotrophic bacteria and ANME were detected at the two sites, suggesting that aerobic and anaerobic methane oxidation are possible in such a niche.ANME often forms consortia with SRB to catalyze sulfate-dependent aerobic methane oxidation(S-DAMO). ANME-1 and ANME-2 are always associated with theDesulfosarcina/Desulfococcuscluster, while ANME-3 is associated with theDesulfobulbusbranch (Boetius et al., 2000; Orphan et al., 2002; Niemann et al., 2006). NoDesulfobulbussequences or minorDesulfococcusandDesulfosarcinasequences were detected in our samples. However,ANME-2a-2b andSEEP-SRB1coexisted in the 8–28-cm layer at site 3400-02 (Figs.9a & 10c), further confirming that AOM can occur at site 3400-02 through the combination of these two groups.
4.5 Potential influence of environmental parameters on the microbial community
Marine microbial communities are aff ected extensively by environmental changes. Many previous studies have focused on the environmental parameters driving the distribution patterns of the microbial community on the surface of the northern Chinese marginal seas (Dong et al., 2014; Xiong et al., 2014; Liu et al., 2015b). Temperature and dissolved oxygen in bottom water have been detected to be vital factors in determining both bacterial and archaeal communities, whereas chlorophyll a in sediment was significant only in structuring archaeal communities in the Chinese marginal seas (Bohai Sea, Yellow Sea, and the northern East China Sea)(Liu et al., 2015b). Shifts in the bacterial community were observed in nitrogen-polluted sediments in the East China Sea (Xiong et al., 2014), indicating that the composition of nutrients is an important factor controlling the microbial community. Few studies have focused on the vertical profile of microbial communities in the northern Chinese marginal seas,with the exception of Qiao et al. (2018)’s study.
In this study, both bacterial and archaeal communities were found to be significantly influenced by sediment depth and TOC (Fig.7). Sediment depth has been suggested a primary factor determining the diff erence in bacterial and archaeal communities in various marine ecosystems (Reed et al., 2006;Devereux et al., 2015; Qiao et al., 2018). We also confirmed in our study that a majority of the identified taxa abundance at the genus level increased or decreased along the sediment depth profile with significant statistical support (Supplementary Fig.S4). Sediment depth may function as a comprehensive proxy of multiple environmental parameters rather than directly aff ecting microbial diversity. The changes in the availability of oxygen or redox state with sediment depth could be the major factor contributing to the stratified distribution of the microbial community. The most abundant archaeal genusCandidatusNitrosopumiluswas abundant in the surface layer and rapidly decreased along the depth profile. This group of ammonia oxidizers has a metabolic trait for aerobic respiration. Most Deltaproteobacteria, includingDesulfatiglansandSEEP-SRB1, belong to strictly anaerobic genera and are sulfate- and sulfur-reducing bacteria, and their relative abundance was very positively correlated with sediment depth. Additionally, microbes preferentially degrade the easily utilizable organic matter in surface sediments, whereas recalcitrant organic matter accumulates in deeper sediments(Cowie and Hedges, 1994; Wakeham et al., 1997).Consequently, microorganisms inhabiting deeper sediments have to meet their metabolic demands by relying on more recalcitrant organic matter (Zonneveld et al., 2010). In this study, the dominant bacterial phylum Chloroflexi and dominant archaeal classes Bathyarchaeia and Lokiarchaeia were believed to have an anaerobic and heterotrophic lifestyle depending on the buried recalcitrant organic matter,probably explaining their higher abundance in the deep sediments (Biddle et al., 2006; Wilms et al.,2006).
The organic matter content is another factor that shapes the bacterial and archaeal communities in marine sediment (Liu et al., 2015b; Oni et al., 2015).At the genus level of the bacterial community,OM60(NOR5) andAqS1affi liated with Gammaproteobacteria andLutimonasandMuriicolaaffi liated with Flavobacteriales preferred substrates with a higher content of TOC (Supplementary Fig.S3). Species withinOM60(NOR5) are aerobic anoxygenic phototrophic bacteria that can use both light and organic compounds as energy sources (Spring et al.,2013). Genomic evidence suggests thatAqS1has a versatile heterotrophic metabolism depending on the organic compounds (Gauthier et al., 2016). The class Flavobacteria is known to play an important role in the recycling of organic carbon in the marine environment (Lozada et al., 2014). They are also known to metabolize high-molecular-weight polymeric substances (Fernández-Gómez et al.,2013). With respect to the archaeal community, we identified that some methanogen genera, includingMethanobacterium,Methanolobus,Methanosarcina,andMethanogenium, were highly negatively correlated with TOC and TN (Supplementary Fig.S3).This phenomenon can be explained by the observation that methanogens cannot use macromolecular organic matter and fail to compete with other microorganisms in sediments with a high organic carbon content.
5 CONCLUSION
Bacterial and archaeal diversity and abundance at site 3500-07 were higher than at 3400-02. Both bacterial and archaeal communities exhibited a clearly stratified distribution due to sediment changes.Archaea Thaumarchaeota and bacteria Gammaproteobacteria and Actinobacteria were higher in the upper sediment layers, whereas the archaea Bathyarchaeia, Lokiarchaeia, and Euryarchaeota, and bacteria Chloroflexi and Deltaproteobacteria were abundant in the deep sediments. Sediment depth and TOC were the vital factors in explaining both the bacterial and archaeal communities. Bacterial communities were abundant in microbes potentially involved in nitrogen and sulfur cycling, while archaeal communities were abundant in microbes potentially involved in ammonia oxidation and methanogenesis.
6 DATA AVAILABILITY STATEMENT
All data generated and/or analyzed during this study are available from the corresponding author upon request.
7 ACKNOWLEDGMENT
We thank all of the scientists and crew members on-board the R/VKexueSanHaofor the assistance provided in the collection of samples and geochemical data during the cruise.
 Journal of Oceanology and Limnology2021年3期
Journal of Oceanology and Limnology2021年3期
- Journal of Oceanology and Limnology的其它文章
- Steady increase in water clarity in Jiaozhou Bay in the Yellow Sea from 2000 to 2018: Observations from MODIS*
- Phylogenetic diversity and bioactivity of culturable deepsea-derived fungi from Okinawa Trough*
- Allelopathic eff ects of mixotrophic dinoflagellate Akashiwo sanguinea on co-occurring phytoplankton: the significance of nutritional ecology*
- Investigation of the decline of Ulva prolifera in the Subei Shoal and Qingdao based on physiological changes*
- Effi ciency of phosphorus accumulation by plankton,periphyton developed on submerged artificial substrata and metaphyton: in-situ observation in two shallow ponds*
- Petroleum exploitation enriches the sulfonamide resistance gene sul2 in off shore sediments