
RON in hepatobiliary and pancreatic cancers: Pathogenesis and potential therapeutic targets
2021-06-05 07:09ShaoLongChenGuoPingWangDanRongShiShuHaoYaoKeDaChenHangPingYao
World Journal of Gastroenterology 2021年20期
Shao-Long Chen, Guo-Ping Wang, Dan-Rong Shi, Shu-Hao Yao, Ke-Da Chen, Hang-Ping Yao
Abstract The receptor protein tyrosine kinase RON belongs to the c-MET proto-oncogene family. Research has shown that RON has a role in cancer pathogenesis, which places RON on the frontline of the development of novel cancer therapeutic strategies. Hepatobiliary and pancreatic (HBP) cancers have a poor prognosis,being reported as having higher rates of cancer-related death. Therefore, to combat these malignant diseases, the mechanism underlying the aberrant expression and signaling of RON in HBP cancer pathogenesis, and the development of RON as a drug target for therapeutic intervention should be investigated. Abnormal RON expression and signaling have been identified in HBP cancers, and also act as tumorigenic determinants for HBP cancer malignant behaviors. In addition, RON is emerging as an important mediator of the clinical prognosis of HBP cancers. Thus, not only is RON significant in HBP cancers, but also RON-targeted therapeutics could be developed to treat these cancers, for example, therapeutic monoclonal antibodies and small-molecule inhibitors.Among them, antibody-drug conjugates have become increasingly popular in current research and their potential as novel anti-cancer biotherapeutics will be determined in future clinical trials.
Key Words: RON; Signal transduction; Hepatobiliary; Pancreatic neoplasms; Molecular targeted therapy
INTRODUCTION
RON receptor tyrosine kinase (RTK; also known as MST1 R) was first identified in 1993 in a cDNA library from human epithelial cells[1 ]. RON belongs to the family of c-MET proto-oncogenes[2 ]. This RTK family only has two members, RON and Met, which share only 34 % overall homology; however, the tyrosine kinase region of the receptors is quite similar at 80 % homology[3 ]. In 1994 , a mouse cDNA was cloned that encoded a homolog of RON, which was termed stem cell-derived tyrosine kinase receptor[4 ].RONis located at human chromosome 3 p21 and this gene shows high conservation in different species, including xenopus, zebrafish, chicken, cats, human, and mouse[4 -11 ]. The RON receptor is initially synthesized as a biologically inactive singlechain precursor (pro-RON), then cleaved into a 145 kDa β-chain and a 35 kDa extracellular alpha chain, which are linked by a disulfide bond, forming the mature receptor. In 1994 , the physiological ligand of RON was identified as macrophagestimulating protein (MSP) [also called hepatocyte growth factor (HGF)-like protein],establishing the MSP-RON signaling system[12 -15 ]. MSP is a member of the plasminogen-related kringle protein family[16 ,17 ]. The humanMSPgene is also located at chromosome 3 p21 and is evolutionarily conserved in different species,similar to RON. The main source of MSP is hepatocytes and MSP circulates in the blood as pro-MSP, which is a biologically inactive single-chain precursor. After subsequent proteolytic conversion, the active mature MSP consists of the disulfidelinked alpha subunit and β-chain. The RON receptor high affinity binding site is in the β-chain and RON activity is regulated by the alpha chain[18 ]. The binding of MSP to RON induces RON dimerization, which activates multiple downstream signaling pathways, leading to RON-mediated cell growth, survival, and invasiveness[19 ,20 ].
In the last two decades, increased research has focused on the tumorigenic and therapeutic roles of RON signaling. Although there have been few studies concerning pathology-related changes in MSP expression, numerous studies regarding aberrant RON activation in various types of tumors have been published, including RON protein overexpression[21 -28 ], oncogenic variant generation[29 -39 ], and persistent activation of downstream signaling pathways[21 -39 ]. In addition, tumorigenic progression and malignancy are associated with functional crosstalk between signaling proteins and RON. In clinical application, increased RON expression can be used for prognostic evaluation of patient survival and disease progression. Hepatobiliary and pancreatic (HBP) cancers have a poor outcome, with high rates of cancerrelated death because of their high incidences of recurrence, metastasis, and invasiveness, and their lack of sensitivity to chemotherapy[40 ]. Complete surgical resection remains the most effective treatment for HBP cancers[40 ]. Among these cancers, the 5 -year survival rate of liver cancer is approximately 30 %, whereas in biliary tract cancer and pancreatic cancer, it is less than 30 % and less than 10 %,respectively[41 ]. The high death rate of pancreatic cancer is caused by the lack of early diagnosis and effective treatment. In pancreatic cancer, most cases are diagnosed when the disease is already at an advanced stage, and only 20 % or less of patients present with potentially curable localized tumors amenable to surgical extirpation[42 ]. Thus,the identification of a novel potential therapeutic strategy is urgently required.Growing evidence suggests a close relationship between HBP cancers and RON dysregulation[24 ,43 ,44 ]. Thus, the present review primarily focuses on the role of RON in the pathogenesis of cancer, especially HBP cancers. Moreover, we summarize the latest progress in the development of strategies targeting RON as potential HBP cancer therapy.
ROLES OF RON AND C-MET IN CARCINOGENESIS
RON and c-MET, both of which are members of the semaphorin family of transmembrane receptor tyrosine kinases, share similar structural and biochemical properties[45 ]. The proteins exist as heterodimers comprising extracellular and transmembrane chains that are linked by disulfide bonds. The RON and c-MET extracellular sequences possess very similar functional domains, including SEMA,which regulates phosphorylation, receptor dimerization, and ligand binding. RON and c-MET are activated by their respective ligands: MSP for RON and HGF for c-MET. c-MET and HGF are expressed in a variety of cell and tissue types. Contrastingly, RON is restricted tightly to epithelial origin cells, whereas liver cells are the major source of its ligand, MSP[46 ]. Independent or ligand-dependent activation of RON and c-MET induces matrix invasion, cell migration, and cell proliferation, all of which are crucial for embryogenesis, wound healing, and tumorigenesis.
Increasing evidence has identified the role of RON and c-Met in the pathogenesis of cancer[47 ]. For example, c-MET and RON overexpression was observed in a variety of primary and metastatic tumors, leading to the activation of aberrant downstream signaling, which contributes to cancer development and progression. Moreover,clinical studies have validated that increased expression of RON and c-MET is a prognostic factor to predict the survival rate and disease progression in certain patients with cancer[48 ,49 ]. Moreover, activation of RON and c-MET promotes a cancer cell malignant phenotype. Increased RON and c-MET expression drives tumor cells to undergo epithelial to mesenchymal transition (EMT), which is characterized by epithelial feature loss and the gain of mesenchymal characteristics[12 ,50 ]. Increased c-MET and RON expression also contributes to acquired chemoresistance[51 ]. Given the above role of the increased expression of c-MET and RON in cancer pathogenesis,targeting RON and c-MET represents a promising cancer therapy strategy.
RON ACTIVATION AND SIGNALING PATHWAY MECHANISMS
Epithelial cells in the skin, adrenal gland, bone, brain, kidney, gut, lung, and liver express low levels of RON[12 ]. The action of RON plays a key role in the motility of epithelial cells, enhancement of adhesion, sperm motility in the epididymis, and embryonic development, as well as the regulation of inflammatory responses[52 ].Under physiological conditions, the main cause of RON activation is stimulation of its ligand, MSP[12 ]. Moreover, three other biochemical events activate RON in tumors:RON overexpression, generation of oncogenic RON variants, and RON transactivation(Figure 1 ). The RON receptor consists of three essential regions: The extracellular domain that recognizes its ligand, the transmembrane domain that anchors the receptor to the membrane, and the intracellular domain that exerts the kinase activity(Figure 1 )[53 ]. The first step for the activation of RON is dimerization at the cell surface, which is caused by the binding of MSP to the extracellular domain containing the specific ligand-binding site, likely resulting in a conformational change in the RON receptor. This activation leads to autophosphorylation at two tyrosine residues(Tyr1238 and Tyr1239 ) located in the A-loop (Phe1227 -Pro1250 ) of the kinase domain.Phosphorylation of these regulatory residues leads to tyrosine kinase function activation, inducing further phosphorylation of residues Tyr1353 and Tyr1360 located in the C-terminal docking site. This then recruits the cytoplasmic molecules growth factor receptor-bound protein 2 (GRB2 ) and Son of Sevenless. In addition, the ubiquitin ligase, casitas B-lineage lymphoma (CBL), binds to the docking site to act as a negative modulator.
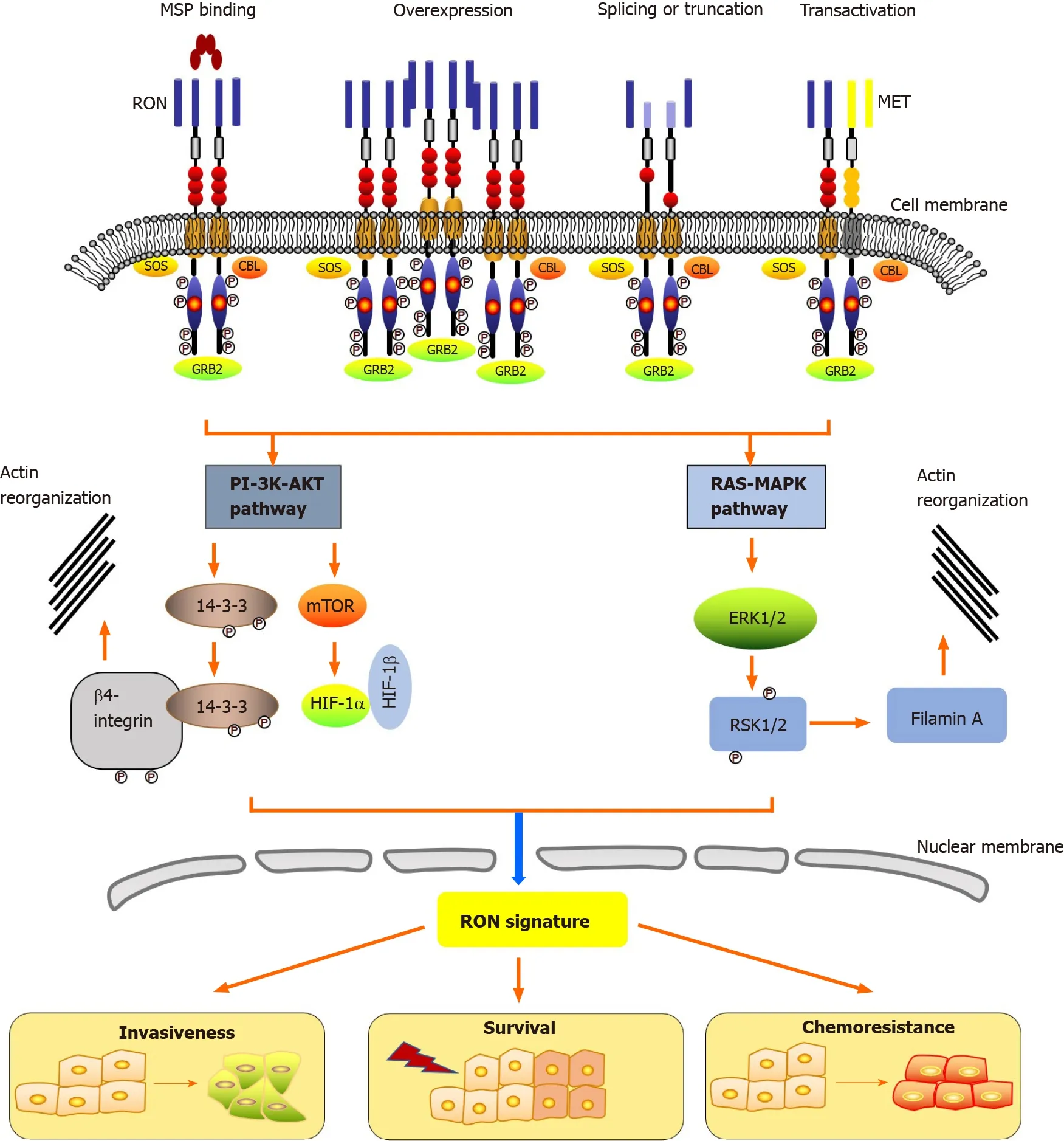
Figure 1 Mechanisms of RON activation and downstream signaling pathways. Classically, macrophage-stimulating protein (MSP) activates RON. In cancer, RON activation is induced by overexpression, splicing or truncation, and transactivation. The RON receptor consists of three regions including the extracellular domain, the transmembrane domain, as well as the intracellular domain. MSP binding to the extracellular domain leads to autophosphorylation of several tyrosine residues in the kinase activation loop or in the C-terminal tail, resulting in the activation of many biological activities, including increased proliferation/survival,motile-invasive activity, and chemoresistance. MSP: Macrophage-stimulating protein; SOS: Son of Sevenless; GRB2 : Growth factor receptor-bound protein 2 ; CBL:Casitas B-lineage lymphoma; 14 -3 -3 : Tyrosine 3 -monooxygenase/tryptophan 5 -monooxygenase activation protein; PI-3 K-AKT: Phosphatidylinositol-4 ,5 -Bisphosphate 3 kinase- protein kinase B; HIF: Hypoxia-inducible factor; RAS-MAPK: RAS-mitogen-activated protein kinase; ERK: Extracellular regulated kinase; RSK: Ribosomal protein S6 kinase; mTOR: Mechanistic target of rapamycin.
The interaction of RON with adaptor proteins, such as β-arrestin-1 and GRB2 ,represents the first step in the bridging of downstream signaling cascades and RON activation.Viaits C-terminal docking site, RON interacts with a variety of cytoplasmic effector molecules, such as phospholipase C gamma, phosphatidylinositol-4 ,5 -Bisphosphate 3 -kinase (PI-3 kinase), Src (SRC proto-oncogene, non-receptor tyrosine kinase), tyrosine 3 -monooxygenase/tryptophan 5 -monooxygenase activation protein(14 -3 -3 ), CBL protooncogene (c-Cbl), heat shock protein family A (Hsp70 ) member 8 (HSC70 ), integrin-β4 , plectin, and protein phosphatase 1 . The classical PI-3 kinaseprotein kinase B (PI-3 K-AKT) and RAS-mitogen-activated protein kinase (RAS-MAPK)pathways are triggered by the interaction of RON’s docking site with downstream signaling proteins. PI-3 K-AKT and RAS-MAPK pathways mediate many biological activities, including increased proliferation and survival, EMT, motile-invasive activity, and chemoresistance. The signaling pathways of RON also play a part in regulating tumorigenic activity. Among them, PI-3 K-AKT and RAS-MAPK pathway coordinated activation plays a crucial role in EMTviaincreased cellular motility[15 ,22 ,54 ,55 ]. Studies using the MDCK cell model showed that EMT mediated by RON is associated with decreased E-Cadherin expression and unregulated vimentin expression, under mediation by RAS-MAPK signaling[56 ,57 ]. The major protein that links EMT to RON signaling is ribosomal protein S6 kinase-2 , which is an intermediate in the MAPK pathway. RON-mediated PI-3 K-AKT signaling is also involved in invasive growth, including increased epithelial cell matrix invasion,migration, and adhesionin vitroand distant metastasis and tumor cell invasionin vivo[54 ,55 ].
ABERRANT RON SIGNALING AND EXPRESSION IN CANCER PATHOGENESIS
In general, normal epithelial cells, including those from the colon, lung, and breast,express low levels of RON; however, cells of a mesenchymal origin do not express RON[12 ,58 ]. RON activation in tumors is frequently the result of receptor overexpression, in contrast to classical MSP binding. Dysregulated RON activation and expression were detected in many types of cancers and have prognostic significance for patient survival. Results from the majority of published studies show that RON expression dysregulation is characterized mainly by elevated expression of wild-type RON and the production of active isoforms, ultimately leading to persistent activation of downstream signaling cascades[12 ]. There have been reports ofRONamplification and point mutation; however, this kind of genetic alteration is observed rarely[26 ]. The relationships between cancer pathogenesis and dysregulated RON signaling and expression were provenviafunctional studies using immunohistochemical (IHC)staining of tumor specimens and cancer cell lines. The first report of wild-type RON overexpression in cancerous tissue was in primary breast cancer samples. Thereafter,IHC staining has further detected wild-type RON in thyroid, bladder, adrenal gland,head and neck, uterus, skin, lung, kidney, pancreatic, colorectal, and other tumors[59 ].These findings are consistent with the results found in other cancers, such as human gliomas, melanoma, and Merkel cell carcinoma, suggesting that aberrant RON expression is also associated with both neurological and skin cancers[18 ]. In breast tissues, the expression of RON is relatively low in normal breast epithelial cells and even in cells from benign lesions (papilloma and adenoma), whereas its was highly expressed in 47 % (35 /75 cases) of histologically varied tumor specimens[25 ]. RON upregulation is associated strongly with its phosphorylation status and invasive activity, suggesting that dysregulated expression of RON functions in human breast carcinoma progression to invasive-metastatic phenotypes. Furthermore, in breast cancer unregulated RON expression was identified as an independent predictor of distant relapse[60 ]. By contrast, in certain tumors, such as hepatocellular carcinoma(HCC), the frequency of wild-type RON expression was relatively low[21 ]; however,its importance remains unknown, although this finding indicates that the wild-type RON is not expressed universally in different tumor types. Moreover, RON overexpression is related to oncogenic RON isoform production, for example RON△160 ,which comprises the deletion of exons 5 and 6 , encoding 109 amino acids in the RON β-chain extracellular sequence[12 ]. RON variants are detected in primary cancer samples and cell lines relatively frequently, and are detected in 40 % to 60 % of cases.Cancer pathogenesis and clinical relevance are likely to be affected by the frequencies and levels of RON isoforms.
Increasing evidence has demonstrated the role of RON in regulating cancer cell invasiveness, which is related to the effects of RON activation on a variety of signaling mechanisms. The activation of complex downstream signaling networks including signal transducer and activator of transcription, β-catenin, JUN N-terminal kinase,MAPK, and PI3 K/AKT pathways are key contributors to RON-mediated aggressive cancer phenotypes. In breast cancer, several signaling pathways that are vital for stemness, invasiveness, and proliferation are activated by RON. For example, the RON-cellular Abelson murine leukemia viral oncogene homolog (c-Abl)-proliferation cell nuclear antigen (PCNA) pathway was identified to contribute to RON-mediated cell growth in breast cancer. Dysregulated RON signaling results in c-Abl activation,consequently leading to PCNA phosphorylation[61 ]. Moreover, in breast cancer, RON signaling regulates the invasiveness of cancer cellviathe activation of the DEK protooncogene (DEK), a DNA-binding non-histone nuclear phosphoprotein that induces closed circular DNA to form positive supercoils[62 ]. This process appears to functionviaa paracrine and autocrine canonical β-catenin signaling loop, which ultimately influences breast cancer stemness. In addition, RON-mediated PI3 K-dependent upregulation of methyl-CpG binding domain 4 , DNA glycosylase (MBD4 ) increases the invasive growth and metastasis of breast cancer cellviathe reprogramming of the DNA methylation of specific target genes[63 ]. Clinical data indicated that in patients with breast cancer, poor prognosis is related to the RON-MBD4 epigenetic pathway[63 ].
RON RECEPTOR AND HEPATOBILIARY CANCERS
In 2030 , it is estimated that more than 1 million people will die because of liver cancer worldwide[64 ]. Primary malignancies of the liver and adjacent biliary tract include HCC, intrahepatic and extrahepatic cholangiocarcinoma (CCA), and gallbladder cancer (GBC). Among them, HCC and intrahepatic CCA account for 85 % and 10 %,respectively[65 ]. Abnormal RON expression has been observed in HCC, which may be related to pathological conditions of this cancer[43 ]. In an HCC cell line study, RON was shown to be associated with oncogenic and invasive phenotypes (e.g., resistance to apoptosis, tumor cell migration, and tumor cell invasion)viaAKT, c-Raf, and extracellular regulated kinase (ERK) signaling cascade modulation[66 ]. Clinically,RON and MET expression in patients with HCC after curative resection suggested no association of RON with overall survival and overall recurrence rates. However,patients with RON+/MET+ disease experienced higher overall recurrence rates compared with those displaying alternative expression patterns[67 ]. Similar to HCC,RON is emerging as an important mediator of CCA pathogenesis and clinical prognosis. Investigation of RON and MET expression in patients with perihilar CCA who underwent histologically curative resection revealed that patients with RON+/MET+ disease showed a worse overall survival rate than patients with other patterns[44 ]. In addition, in patients with extrahepatic CCA, the complete loss of MET,RON, or both (and their overexpression) was a factor for poor prognosis, likely due to the high rate of lymph-node metastasis[68 ]. Recently, Cheng and co-workers indicated that BMS-777607 , a MET-RON dual inhibitor, inhibited HuCCT1 and KKU-100 human CCA cell growth, and decreased the growth of tumors in CCA rats. They further found that for patients with CCA who had previously undergone hepatectomies, upregulation of RON and MET was a predictor of poor survival[69 ]. Taken together, these studies suggest that the aberrant RON expression found in human hepatobiliary cancer samples and cell lines is closely related to pathological conditions and clinical outcome.
RON RECEPTOR AND PANCREATIC CANCER
The majority of malignant neoplasms of the pancreas are adenocarcinomas, among which pancreatic ductal adenocarcinoma is the most common malignancy, representing an excess of 95 % of all pancreatic malignancies[70 ]. Pancreatic cancer presents a substantial health problem and is associated with an extremely poor prognosis because of the non-specific symptoms in patients, its aggressive and remarkable resistance to most conventional treatment options, and the fact that it harbors multiple genetic and epigenetic alterations[42 ]. Therefore, novel therapies to treat pancreatic cancer are urgently required. In recent years, the function of RON in pancreatic cancer has been identified extensively in a variety of model systems, such as animal, cellular,and clinical settings. To date, researchers have reported that RON is expressed in various pancreatic cancer cell lines, such as CFPAC-1 , ASPC-1 , Hs766 .T, L3 .6 pl,HPAFII, HPAC, Capan-2 , and BXPC-3 . However, MIA-PACA-2 cells show minimal RON expression[71 ]. The association of RON with Kras-driven pancreatic carcinogenesis was investigated using genetically engineered mouse models. The results showed that overexpression of RON accelerated pancreatic intraepithelial neoplasia(PanIN) progression, enhanced acinar-ductal metaplasia, and promoted tumor progression towards invasive pancreatic cancer[72 ]. Moreover, the study proved that the initiation of PanIN was slowed by RON kinase domain genetic inactivation,resulting in smaller tumors, and eventually prolonging tumor-bearing mouse survival[72 ]. Great progress has been made in our understanding of the clinical relevance of RON in pancreatic cancer, which has focused mainly on RON expression status in pancreatic cancer samples and its possible utility as a prognostic biomarker for patient survival. IHC staining using anti-RON antibodies is a commonly used approach to evaluate RON expression in various experimental settings. Several studies have identified positive sample rates in pancreatic cancer specimens such as 70 %, 88 %,96 %, 80 %, and 86 %, respectively[21 ,73 ]. Meanwhile, in pancreatic cancer samples, high RON expression has been detected, whereas minimal levels were detected in their corresponding normal epithelial cells. Notably, during pancreatic cell tumorigenic progression, the frequency and level of RON expression increased[22 ,74 ]. Among human pancreatic tissue samples, RON expression was detected in 83 % of metastatic lesions, 79 % of primary lesions, and 93 % of high-grade PanIN using immunohistochemistry, with low expression being detected in low-grade PanIN and normal ducts (18 % and 6 %, respectively), suggesting that RON might function in pancreatic carcinogenesis and metastatic progression[22 ]. Moreover, RON expression levels were significantly related to overall survival in patients with pancreatic cancer, indicating that RON could be an important indicator of prognosis in pancreatic cancer[75 ].Conflicting results between RON expression and pancreatic cancer prognosis were found in an early study[76 ], thus more research is needed to determine the utility of RON as a prognostic biomarker in patients with pancreatic cancer.
Primary and metastatic pancreatic tumor specimens and high grade PanIN lesions show increased RON expression[22 ]. Accumulating evidence suggests that dysregulated RON signaling and activation might function in tumor formation and metastasis. Generally, activation of RON results in increased pancreatic cancer tumorigenic stemness, chemoresistance, survival capability, angiogenesis, and cell invasiveness[73 ]. Among them, invasiveness occursviaa phenotype resembling EMT.A study found that MSP treatment of the pancreatic cancer cell line L3 .6 pl resulted in increased cell invasion, cell migration, and ERK phosphorylation[24 ]. Activation of RON resulted in decreased levels of membrane-bound E-cadherin together with βcatenin nuclear translocation, which resembled EMT. Treated L3 .6 pl cells acquired a spindle shape and lost their polarity, their intercellular separation increased, and more pseudopodia were formed[24 ]. Aberrant RON activation in collaboration with other growth factors, such as transforming growth factor-β, contributes to the phenotypic changes of pancreatic cancer cells towards EMT. Additionally, an investigation of RON signaling-mediated angiogenesis regulation in pancreatic cancer found that RON signaling leads to MAPK-mediated pancreatic cancer cell production of the wellcharacterized angiogenic protein, vascular endothelial growth factor. RON activation also caused the promotion of microtubule formation[77 ]. Finally, the RON signaling pathway also plays a part in chemoresistance, which is associated with enhanced survival capability[51 ,78 ]. Short hairpin RNA (shRNA)-mediated silencing ofRONexpression in pancreatic cancer xenografts resulted in increased sensitivity to gemcitabine therapy and susceptibility to apoptosis[51 ]. In the light of the above findings, it is clear that RON signaling is crucial for pancreatic cancer formation and metastasis.
RON AS A THERAPEUTIC TARGET FOR HBP CANCERS
Based on the pathogenic role of RON in cancers, including HBP cancers, efforts have focused predominantly on establishing RON as a drug target for therapeutic intervention[73 ]. A variety of techniques were proposed to effectively block RON signaling and expression. One approach is to inhibit RON expression using gene silencing with small interfering RNAs (siRNAs). In pancreatic cancer xenografts,RONsilencing caused growth inhibition by enhancing their apoptosis susceptibility andviasensitization to gemcitabine therapy[51 ]. Thus, delivery of RON-specific siRNAs could have therapeutic potential. In addition, small-molecule kinase inhibitors (SMKIs),which block the receptor tyrosine kinase domain eithervianon-competitive inhibition orviaATP competition, have been proposed[45 ]. The structural similarities between the kinase domains of MET and RON resulted in the development of selective small molecule inhibitors targeting both the RON and MET kinase domains, with slightly different IC50 values. As described above, BMS-777607 , a MET-RON dual inhibitor, has shown its effects in inhibiting the growth of human intra-hepatic CCA cell lines and also decreasing tumor growth in intrahepatic CCA rats[69 ]. However, preclinical studies to prove RON as a drug target showed unsatisfactory results when using RON-specific SMKIs[46 ,79 -81 ]. The first reason for the above result is that HBP cancer cell survival does not depend on RON signaling. Second, an SMKI that specifically inhibits only RON kinase activities is not available. Synthetic SMKIs, including Tivantinib, BMS-777607 , INCB28060 , Compound-1 , and PHA665752 , all recognize both RON and MET, with similar kinase-binding affinities[73 ]. Thus, the characterized SMKI RON or MET-specific inhibitors are actually multiple RTK inhibitors and the
development of SMKIs that exclusively target RON has been a challenge.
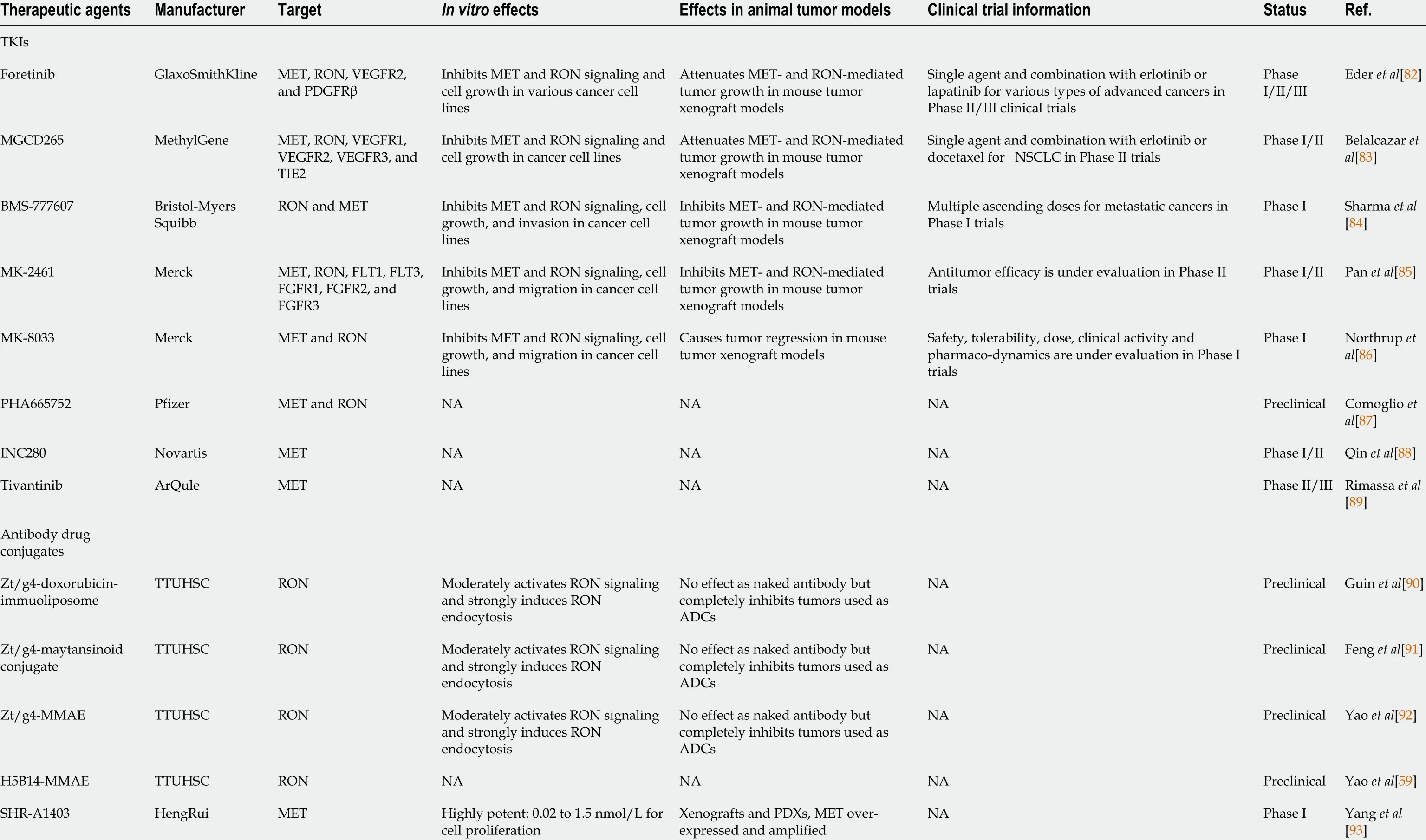
Table 1 Tyrosine kinase inhibitors and antibody drug conjugates specific to c-MET and RON
A more realistic approach is using anti-RON therapeutic monoclonal antibodies(TPABs) to treat HBP cancers. For instance, anti-RON antibody Zt/c9 -directing doxorubicin-immunoliposomes was effective at killing purified pancreatic cancer stem cellsin vitro. The underlying mechanism is that Zt/c9 -directing doxorubicin-immunoliposomes specifically interact with pancreatic cancer stem cells and rapidly cause RON internalization, which leads to the uptake of liposome-coated Dox. In addition,preclinical models have been constructed using anti-RON TPABs, such as 7 G8 , 6 D4 ,6 E6 , narnatumab (or IMC-RON8 ), Zt/f2 , and IMC-41 A10 , which either block MSP binding by recognizing RON’s ligand-binding pocket or affect receptor dimerization by interacting with RON’s extracellular domain (e.g., SEMA), thereby attenuating signaling transduction[73 ]. However, previous studies concerning TPAB therapy revealed only partial inhibition of tumor growth, and there have been no reports of single anti-RON TPAB administration achieving complete inhibition. Thus, strategies to maximize anti-RON TPABs’ therapeutic activity have moved on to an exciting new area. Anticancer therapeutic agents comprising antibody-drug conjugates (ADCs)combine the specificity of antibodies with the high potency of cytotoxins to enhance cell killing[12 ]. To generate RON-targeted ADCs, the anti-RON monoclonal antibodies PCM5 B14 and Zt/g4 were selected to prepare immunotoxins. To generate Zt/g4 and PCM5 B14 -based ADCs, cytotoxic payloads with different mechanisms of action were conjugated, including pyrrolobenzodiazepine, duocarmycin (DCM), monomethyl auristatin E, and maytansinoid derivative 1 , forming for example, Zt/g4 -MME and PCM5 B14 -DCM[59 ]. Preclinical studies identified Zt/g4 - and PCM5 B14 -based ADCs as lead candidates for clinical development and increased the chance of their entering into clinical trials (Table 1 ).
CONCLUSION
RON was identified over two decades ago, and since then, accumulating evidence has indicating RON’s involvement in tumorigenesis, which has resulted in increased momentum for developing RON as a target for therapeutic drug intervention. As outlined in this review, the identification of dysregulated activation and expression of RON in various cancers has expanded our understanding of the mechanisms underlying cancer pathogenesis. Importantly, HBP cancers are characterized pathologically by the dysregulated signaling and expression of RON, which also act as tumorigenic determinants for the malignant behavior of HBP cancers. Moreover,abnormal RON expression is important to determine the clinical outcome of patients with HBP cancers. The growing knowledge concerning the crucial role of RON in HBP cancers can be translated into promising cancer therapeutic strategies. Consequently, a number of clinical trials are underway to assess SMKIs and TPABs targeting RON as a molecular target, some of which have shown promising results. Furthermore,PCM5 B14 - and Zt/g4 -based ADCs, as anti-RON ADCs, are receiving increased research interest and the striking advances in exploiting anti-RON ADCs will hopefully translate into clinical treatments for patients with HBP cancer in the future.
 World Journal of Gastroenterology2021年20期
World Journal of Gastroenterology2021年20期
- World Journal of Gastroenterology的其它文章
- Deep learning for diagnosis of precancerous lesions in upper gastrointestinal endoscopy: A review
- State of machine and deep learning in histopathological applications in digestive diseases
- COVID-19 in normal, diseased and transplanted liver
- Upregulation of long noncoding RNA W42 promotes tumor development by binding with DBN1 in hepatocellular carcinoma
- Development and validation of a prognostic model for patients with hepatorenal syndrome: A retrospective cohort study
- Inflammatory bowel disease in Tuzla Canton, Bosnia-Herzegovina: A prospective 10 -year follow-up