
六氢吡咯吲哚生物碱Esermethole的全合成
2021-06-04 08:51彭天凤沈先福奚永开周永云李淑敏陈立翔王云肖雷兴富赵静峰
云南大学学报(自然科学版) 2021年3期
彭天凤,沈先福,**,奚永开,周永云,李淑敏,陈立翔,杨 博,王云肖,雷兴富,赵静峰**
(1.曲靖师范学院生物资源与食品工程学院,化学与环境科学学院,云南 曲靖655011;2.云南大学云南省天然产物转化与利用及教育部自然资源药物化学重点实验室,云南 昆明650091)
六氢吡咯吲哚是吲哚类生物碱天然产物中关键结构单元,组成了一些结构多样和独特生物学合成途径的天然产物[1-2].具有吡咯吲哚啉结构单元的生物碱,广泛存在于自然界天然产物中,并表现出丰富多样的生物活性,如抗菌和抗癌活性[3-6].例如,Physostigmine作为吲哚环C3a位甲基取代天然产物,最初从非洲卡拉巴豆植物中分离得到,是一种潜在的乙酰-和丁酰胆碱酯酶可逆抑制剂,在临床上用来治疗青光眼、重症肌无力和阿尔茨海默病[7].然而,由于其进入生物体作用时间短、低生物利用度以及治疗范围狭窄,其临床使用受到了严格的限制.Phenserine从结构上来说,是天然产物Physostigmine的类似物,作为Physostigmine临床药物的替代者,因为具有抑制乙酰胆碱酯酶以及β-淀粉样斑块在大脑中沉积能力的生物活性受到广泛关注[8-9].六氢吡咯吲哚类生物碱具有独特和新颖的化学结构,它们拥有吲哚环C3a位全碳季碳手性中心,其合成的难点和主要挑战是立体选择性构筑其C3a位全碳季碳手性中心.由于它们良好的生物活性和独特的化学结构,促使合成化学工作者开发简洁、高效且具有创造性的方法,制备它们结构中的核心杂环骨架,这已成为近年来化学合成领域研究热点,一系列新颖合成该类生物碱的方法被陆续报道出来[10-21].具有代表性的六氢吡咯生物碱结构见图1.2011年,张洪彬等[17]基于钯催化邻溴苯胺类化合物芳基化串联烷基化反应,构建了含季碳中心的氧化吲哚中间体,通过后续的化学转化,进而完成了六氢吡咯吲哚生物碱Esermethole的合成.2012年,Garg等[18]报道了一种基于中断的Fischer吲哚环化反应,构建了吡咯并二氢吲哚关键结构,进而完成了天然产物Esermethole的合成.2015年,祝介平等[14]报道了一种基于金属钯催化Heck/分子内碳-氢活化的串联反应,实现了天然产物(+)-Esermethole的合成.近期,苏陈良等[20]发展了一种基于光催化碳-氢活化的方法,实现了天然产物Esermethole的合成.通过文献调研,目前天然产物Esermethole大部分合成工作,大多是以取代吲哚或色氨酸衍生物作为起始原料,只有很少一部分合成路线是以非吲哚或非氧化吲哚为起始原料[2-4],这对于得到结构多样的六氢吡咯吲哚生物碱天然产物,具有一定局限性.基于前期铜催化的串联反应[22-24],我们报道一条新颖的天然产物Esermethole全合成路线.
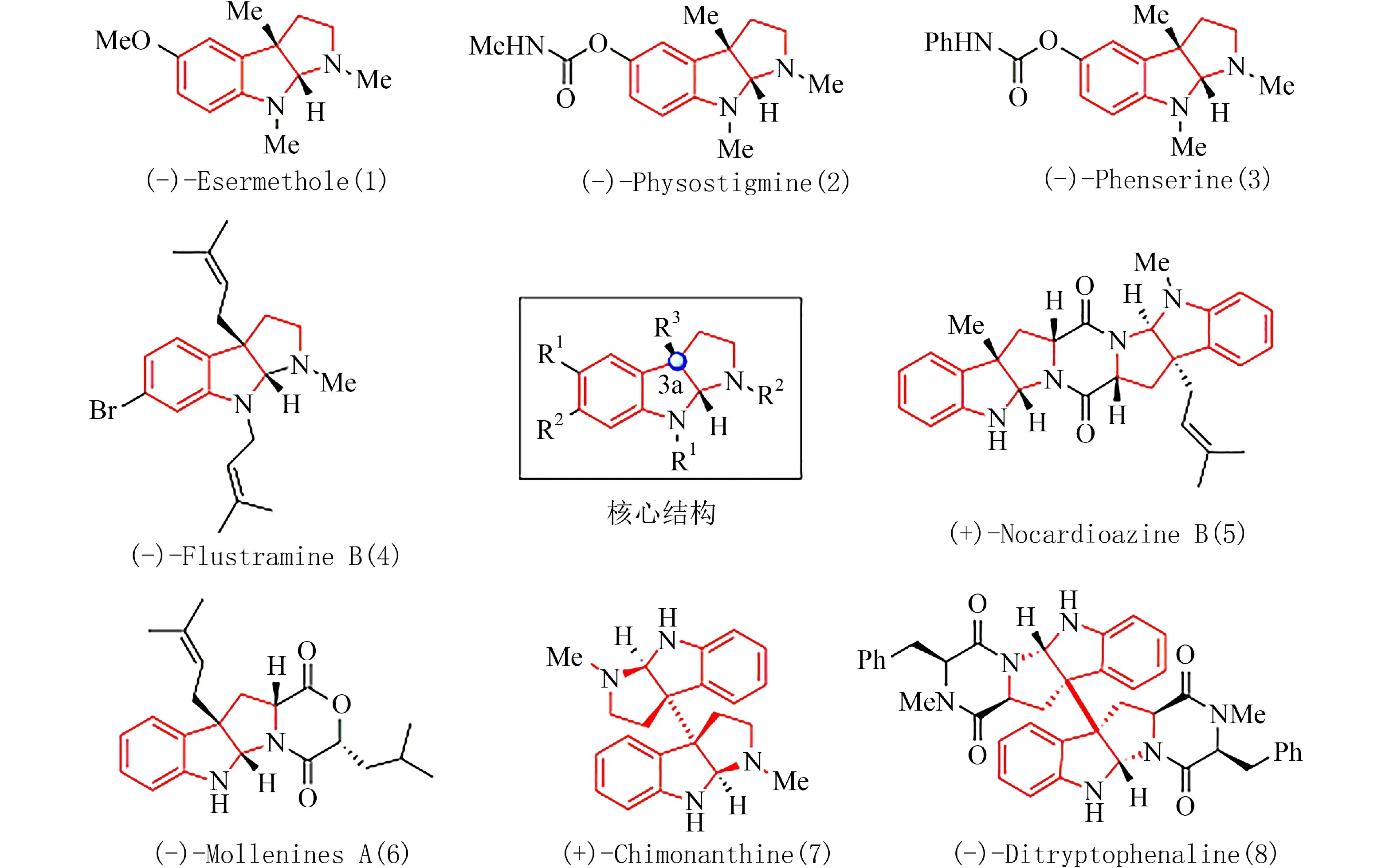
图1 具有代表性的六氢吡咯生物碱Fig.1 Representative pyrroloindoline alkaloids
1 天然产物Esermethole的逆合成分析
天然产物Esermethole逆合成分析路线如图2所示.通过合理的构思,我们设想天然产物Esermethole可以分别由关键氧化吲哚化合物9水解脱除叔丁基亚磺酰基辅基、还原氨化环化得到;而氧化吲哚化合物9可由酰胺10经过铜催化邻溴苯胺化合物芳基化串联烷基化反应[22]得到;酰胺化合物10则是由亚胺11经过亚胺还原、氮烷基化得到;醇12经过氧化、与S型叔丁基亚磺酰胺缩合,就可以得到亚胺11;醇化合物12则是由苯胺类化合物13与γ-丁内酯14通过氨解开环、氮烷基化得到.
2 实验部分
2.1 仪器与试剂Bruker AV-400 MHz核磁共振仪(以CDCl3为溶剂,TMS为内标);Thermo Scientific Q Exactive高分辨率质谱仪(ESI).本实验所用试剂均为市售分析纯.XT4A显微熔点测定仪(北京科仪电光仪器厂);薄层色谱层析硅胶:GF254(青岛海洋化工厂分厂);柱层析硅胶:0.0 54~0.0 77 mm(青岛海洋化工厂).IR在Bio-Rad FTS-135型红外光谱仪上测定;熔点由国产北京泰克仪器设备有限公司生产的XT-4型双目显微熔点测定仪测定,温度计未校正;洗脱剂采用乙酸乙酯、石油醚(b.p.60~90℃)、二氯甲烷、甲醇等;所用其他溶剂均用标准方法纯化后使用;高效液相色谱(HPLC)由大赛璐手性柱和Agilent 1200液相色谱仪检测,使用正己烷与异丙醇作为流动相.

图2 Eser methole的逆合成分析Fig.2 Retrosynthetic analysisof Esermethole

图3 Eser methole的全合成路线Fig.3 Total synthesisof Esermethole
2.2 天然产物Esermethole的全合成首先,从商品化原料2-溴-4-甲氧基苯胺13和γ-丁内酯14出发,如图3所示,在三甲基铝(AlMe3)的作用下,γ-丁内酯14经过氨解开环反应,以96%反应产率,得到酰胺15;然后15在碳酸铯以及碘甲烷为烷基化试剂的条件下,发生酰胺氮烷基化反应,以94%反应产率,得到了氮甲基化产物12.随后将醇化合物12用戴斯马丁氧化剂(DMP)氧化,醇氧化为醛,以85%反应产率,得到化合物醛16;然后进一步制备含手性硫的亚胺化合物11,在四乙氧基钛为脱水剂、催化剂以及四氢呋喃为反应溶剂条件下,醛16与手性S型叔丁基亚磺酰胺顺利地发生脱水缩合反应,以92%的反应产率,得到亚胺化合物11,接着亚胺化合物11经过硼氢化钠(NaBH4)还原,以90%产率,得到亚胺还原产物17,进而17在碘甲烷(CH3I)为烷基化试剂、氢化钠(NaH)作碱的反应条件下,叔丁基亚磺酰胺中的氮发生甲基化反应,以87%产率,得到了铜催化前体化合物10.然后,我们尝试铜催化芳基化串联烷基化的关键反应,在碘化亚铜(CuI)/双三甲基硅基胺基锂[LiN(SiMe3)2]以及碘甲烷(CH3I)为烷基化试剂一锅法反应条件下,邻溴苯胺化合物10顺利发生铜催化芳基化串联烷基化关键反应,以50%的反应产率以及dr=81.5 ∶18.5 的非对映选择性,得到了关键含有芳基全碳季碳中心的氧化吲哚中间体9和9′,该反应能顺利放大到克级规模,反应的产率不受影响.唯一遗憾的是,得到的非对映异构体9和9′不能通过硅胶柱层析分离.然后我们继续完成天然产物Esermethole的后续合成步骤,氧化吲哚化合物9和9′经过稀盐酸脱除分子中的叔丁基亚磺酰基辅基,以91%的产率,得到了氨基化合物18;最后,化合物18在二异丁基铝氢(DIBAL-H)的还原作用下,顺利地发生还原氨化环化反应,以88%的产率,得到了六氢吡咯吲哚生物碱Esermethole. 通过与已知文献中的核磁共振氢谱、碳谱谱图数据对比[14],我们发现得到的天然产物的核磁共振氢谱、碳谱谱图与已知报道的天然产物谱图一致.最终,我们以9步反应以及22.1%的总产率,完成了天然产物Esermethole的全合成,且该合成路线中前面8步反应,能以克级甚至十几克级规模进行,扩大反应规模,不影响反应的产率,这样就可以大量制备天然产物Esermethole及其类似物,为相关中间体化合物的活性研究提供基础.然后参照已知文献[12,18],也可以实现天然产物Physostigmine和Phenserine的形式合成.
2.3 化合物的合成
2.3.1 N-(2-溴-4-甲氧基苯基)-4-羟基丁酰胺(15)的合成 称取2-溴-4-甲氧基苯胺(20.1g,100.0 mmol)于500 mL三口圆底烧瓶中,加入300 mL无水甲苯,氮气保护下,脱气3次,然后缓慢加入三甲基铝(2.0 mol/L,60 mL),室温搅拌反应30 min,再缓慢加入1,4-丁内酯(9.2 mL,120.0 mmol),室温下搅拌反应24 h.反应完全后,在冰水浴下,缓慢加入稀盐酸(1 mol/L,360 mL),继续搅拌1.5 h,乙酸乙酯萃取(400 mL×3),合并有机相,并用200 mL饱和食盐水洗涤,无水硫酸钠干燥.减压浓缩,将浓缩后得到的粗产品,经柱层析分离纯化[V(石油醚)∶V(乙酸乙酯)=1∶3]得到白色固体15(27.5 g,96.0 mmol),收率96%. m.p. 67~69℃.1H NMR(400 MHz,CDCl3)δ:8.00(s,1H),7.67(d,J=9.0 Hz,1H),6.96(d,J=2.8 Hz,1H),6.70(dd,J=9.0,2.8 Hz,1H),4.02~3.90(m,1H),3.67(s,3H),3.64~3.55(m,2H),2.44(t,J=7.2 Hz,2H),1.9 1~1.7 9(m,2H).13C NMR(100 MHz,CDCl3)δ:172.2 3,156.9 0,128.5 5,125.3 0,117.4 0,116.6 2,113.5 1,61.4 5,55.5 2,33.8 7,28.1 2.FTIR(KBr,thin film,cm-1)ν:3 268,2 941,1662,1 603,1520,1495,1 278,1 218,1 035,750.HRMS(ESI-TOF)m/z([M+H]+,C11H15BrNO3)计算值288.023 0,实验值288.0226.
2.3.2 N-(2-溴-4-甲氧基苯基)-4-羟基-N-甲基丁酰胺(12)的合成 称取化合物15(22.9 g,80.0 mmol),加入无水乙腈和无水N,N二甲基甲酰胺[V(CH3CN)∶V(DMF)=2∶1,300 mL],再加入碳酸铯(39.0 g,120 mmol)和碘甲烷(7.5 mL,120.0 mmol),室温反应5 h.然后用垫有硅藻土的抽滤漏斗抽滤,乙酸乙酯(150 mL×3)洗涤,合并有机相减压浓缩,经柱层析纯化[V(石油醚)∶V(乙酸乙酯)=1∶3]得到无色油状液体12(22.7 g,75.4 mmol),收率94%.1H NMR(400 MHz,CDCl3)δ:7.16(d,J=1.6 Hz,1H),7.15(d,J=4.2 Hz,1H),6.87(dd,J=8.7,2.8 Hz,1H),3.79(s,3H),3.62~3.47(m,2H),3.32~3.20(brs,1H),3.12(s,3H),2.13~2.04(m,2H),1.84~1.70(m,2H).13C NMR(100 MHz,CDCl3)δ:174.07,159.75,135.25,129.97,123.63,118.75,114.83,62.52,55.87,36.22,31.53,27.75.FTIR(KBr,thin film,cm-1)ν:3 418,2939,1646,1 496,1287,1 034,849,746. HRMS(ESI-TOF)m/z([M+H]+,C12H17BrNO3)计算值302.038 7,实验值302.038 6.
2.3.3 N-(2-溴-4-甲氧基苯基)-N-甲基-4-乙酰基乙酰胺(16)的合成 称取化合物12(18.1 g,60 mmol),加入200 mL无水二氯甲烷溶解,冰水浴搅拌下,加入碳酸氢钠(10.1 g,120 mmol)后,再缓慢戴斯马丁氧化剂(27.9 g,66 mmol),反应2 h后用垫有硅藻土抽滤漏斗抽滤,乙酸乙酯(200 mL×3)洗涤,滤液减压浓缩,经柱层析纯化[V(石油醚)∶V(乙酸乙酯)=1∶2]得到淡黄色油状液体16(15.2 g,50.8 mmol),收率85%.1H NMR(400 MHz,CDCl3)δ:9.71(s,1H),7.20(d,J=8.7 Hz,1H),7.13(d,J=2.8 Hz,1H),6.86(dd,J=8.7,2.8 Hz,1H),3.77(s,3H),3.10(s,3H),2.82~2.70(m,1H),2.65~2.52(m,1H),2.36~2.23(m,1H),2.20~2.09(m,1H).13C NMR(100 MHz,CDCl3)δ:201.06,171.48,159.75,134.90,130.06,123.61,118.69,114.80,55.77,38.78,36.02,26.6 7.FTIR(KBr,thin film,cm-1)ν:2 941,2839,1719,1 660,1 496,1 288,l034,850.HRMS(ESITOF)m/z([M+H]+,C12H15BrNO3)计算值300.023 0,实验值300.023 0.
2.3.4 (S,E)-N-(2-溴-4-甲氧基苯基)-4-(叔丁基亚磺酰亚胺基)-N-甲基丁酰胺(11)的合成 称取化合物16(15.0 g,50 mmol),加入200 mL无水四氢呋喃溶解,再加入(S)-叔丁基亚磺酰胺(12.1 g,100 mmol)和钛酸四乙酯(22.8 g,100 mmol),氮气保护下脱气3次,然后在60℃油浴加热下,反应12 h.反应完全后,加入100 mL饱和食盐水溶液,剧烈搅拌1 h,然后用垫有硅藻土抽滤漏斗抽滤,乙酸乙酯(200 mL×3)洗涤,减压浓缩,粗产品经柱层析分离纯化[V(石油醚)∶V(乙酸乙酯)=1∶2]得到无色粘状液体11(18.4 g,45.7 mmol),收率92%.1H NMR(400 MHz,CDCl3,compound exists as a mixture of rotamers)δ:8.01(q,J=3.3 Hz,1H),7.18~7.13(m,2H),6.90~6.83(m,1H),3.79(s,3H),3.12(s,1.5H),3.11(s,1.5H),2.96~2.60(m,2H),2.44~2.13(m,2H),1.13(s,4.5H),1.11(s,4.5H).13C NMR(100 MHz,CDCl3,compound exists as a mixture of rotamers)δ:171.42,168.31,168.14,159.82,159.79,135.21,135.13,130.12,130.08,123.89,123.78,118.77,118.75,114.92,114.88,56.91,56.77,55.87,36.09,36.04,31.36,31.34,29.42,29.03,22.38,22.35.FTIR(KBr,thin film,cm-1)ν:3 398,2974,2901,1 664,1 624,1 496,1 287,1079,1047.HRMS(ESI-TOF)m/z([M+H]+,C16H24BrN2O3S)计 算 值403.0 68 6,实验值403.0 68 9.
2.3.5 (S)-N-(2-溴-4-甲氧基苯基)-4-(叔丁基亚磺酰胺基)-N-甲基丁酰胺(17)的合成称取化合物11(16.1 g,40 mmol),加入200 mL无水甲醇,然后在0℃冰水浴下,缓慢地加入硼氢化钠(4.54 g,120 mmol),继续反应5 h.反应完全后,在冰水浴下缓慢加入80 mL饱和的氯化铵溶液,室温下搅拌1 h,然后加入150 mL水稀释,减压浓缩,乙酸乙酯萃取(200 mL×3),无水硫酸钠干燥,滤液减压浓缩,将浓缩后得到的粗产品,经柱层析纯化[V(石油醚)∶V(乙酸乙酯)=1∶2]得到淡黄色油状液体17(14.5 g,35.9 mmol),收率90%.1H NMR(400 MHz,CDCl3,compound exists as a mixture of rotamers)δ:7.22~7.09(m,2H),6.93~6.83(m,1H),3.82(s,3H),3.49~3.37(m,0.5H),3.36~3.27(m,0.5 H),3.2 2~3.1 0(m,4H),3.0 8~2.9 3(m,1H),2.1 0~1.9 5(m,2H),1.8 9~1.7 8(m,2H),1.1 5(s,9H).13C NMR(100 MHz,CDCl3,compound exists as a mixture of rotamers)δ:172.9 3,172.7 8,159.8 2,135.4 5,130.2 6,130.1 0,123.9 1,123.8 0,118.8 3,114.9 1,114.8 7,55.9 3,55.6 9,55.6 8,45.2 5,45.0 6,36.1 4,36.1 1,31.3 6,31.1 8,26.2 2,26.1 8,22.7 5,22.7 4.FTIR(KBr,thin film,cm-1)ν:3445,3 243,2 956,1 655,1496,1 287,1036,850,602. HRMS(ESI-TOF)m/z([M+H]+,C16H26BrN2O3S)计算值405.0842,实验值405.0843.
2.3.6 (S)-N-(2-溴-4-甲氧基苯基)-4-(叔丁基亚磺酰甲基胺)-N-甲基丁酰胺(10)的合成 称取化合物17(12.1 g,30 mmol),然后加入150 mL无水四氢呋喃溶解,冰水浴下,缓慢加入氢化钠(60%,1.8 g,45 mmol),再加入碘甲烷(2.8 mL,45 mmol),缓慢恢复至室温反应12 h.反应完后缓慢加入100 mL饱和氯化铵溶液,搅拌0.5 h后,乙酸乙酯萃取(200 mL×3),无水硫酸钠干燥,滤液减压浓缩,将浓缩后得到的粗产品,经柱层析分离纯化[V(石油醚)∶V(乙酸乙酯)=1∶2]得到淡黄色油状液体10(10.9 g,26.0 mmol),收率87%.1H NMR(400 MHz,CDCl3,compound exists as a mixture of rotamers)δ:7.19~7.12(m,1H),6.90~6.86(m,1H),3.81(s,3H),3.14(s,3H),3.05~2.85(m,2H),2.57(s,1.5H),2.56(s,1.5H),2.05~1.88(m,2H),1.87~1.77(m,2H),1.09(s,4.5H),1.08(s,4.5H).13C NMR(100 MHz,CDCl3,compound exists as a mixture of rotamers)δ:172.56,159.82,135.44,135.42,130.14,123.88,118.83,114.90,114.88,58.26,55.93,54.03,53.80,36.07,32.83,32.71,31.16,31.11,23.87,23.56.FTIR(KBr,thin film,cm-1)ν:3 465,2 968,1662,1496,1 442,1 287,1 068,1037.HRMS(ESI-TOF)m/z([M+H]+,C17H28BrN2O3S)计 算 值419.0 99 9,实验值419.1 00 3.
2.3.7 (S)-N-(2-((S)-5-甲氧基-1,3-二甲基-2-氧化吲哚啉-3-乙基)-N,2-二甲基丙烷-2-亚磺酰胺(9)和(S)-N-(2-((R)-5-甲氧基-1,3-二甲基-2-氧化吲哚啉-3-乙基)-N,2-二甲基丙烷-2-亚磺酰胺(9′)的合成 在250 mL双口圆底瓶中,称取碘化亚铜(CuI,95.0 mg,0.5 mmol)和化合物10(2.2 g,5.0 mmol),加入100 mL无水四氢呋喃(先用氢化锂铝回流加热处理,然后用金属钠回流处理得到),高纯氮气保护下脱气3次,室温搅拌下加入双三甲硅基胺基锂(1.0 mol/L in THF,10 mL),再脱气3次,然后在80℃油浴加热下,反应8 h,然后停止加热,恢复至室温,再冷却至0℃,加入碘甲烷(0.3 mL,5 mmol)反应2 h.反应完后加入10 mL饱和氯化铵溶液搅拌30 min,然后加入30 mL水,乙酸乙酯萃取(100 mL×3),无水硫酸钠干燥,滤液减压浓缩,将浓缩后得到的粗产品,经柱层析纯化[V(石油醚)∶V(乙酸乙酯)=1∶2]得到淡黄色油状液体9和9′(880 mg,2.5 mmol),收率50%.1H NMR(400 MHz,CDCl3,compound exists as a mixture of rotamers)δ:6.82~6.76(m,2H),6.75~6.70(m,1H),3.7 9(s,1H),3.7 8(s,2H),3.1 7(s,3H),2.7 8~2.5 7(m,2H),2.5 5(s,2H),2.5 3(s,1H),2.2 8~2.1 5(m,1H),2.0 1~1.8 9(m,1H),1.3 5(s,3H),1.0 9(s,3H),1.0 8(s,6H).13C NMR(100 MHz,CDCl3,compound existsas a mixture of rotamers)δ:179.5 0,156.3 5,156.3 2,136.8 1,136.7 3,134.4 7,134.4 5,112.1 8,112.0 4,110.3 3,110.2 1,108.6 6,58.1 3,58.0 3,55.9 6,49.9 5,49.4 8,47.3 3,47.3 1,36.5 5,36.4 5,33.7 5,33.2 2,26.4 3,26.4 1,24.4 1,24.3 8,23.5 1,23.4 5.FTIR(KBr,thin film,cm-1)ν:3467,2 960,1 704,1600,1 496,1 469,1 290,1 124,1 065,1034.HRMS(ESI-TOF)m/z([M+H]+,C18H29N2O3S)计算值353.189 4,实验值353.1 894.
2.3.8 (S)-5-甲氧基-1,3-二乙基-3-(2-甲氨基乙基)-吲哚啉-2-酮(18)的合成 称取化合物9和9′(1.4 g,4.0 mmol),然后加入40 mL无水甲醇溶解,再加入HCl(2 mol/L,4.0 mL),室温下反应2 h,然后加入饱和的碳酸氢钠溶液中和至弱碱性,再加入15 mL水,二氯甲烷(200 mL×3)萃取,无水硫酸钠干燥,滤液减压浓缩,将浓缩后得到的粗产品,经柱层析纯化[V(二氯甲烷)∶V(甲醇)∶V(氨水)=300∶10∶1]得到淡黄色油状液体18(862 mg,3.5 mmol),收率87%.1H NMR(400 MHz,CDCl3)δ:6.81~6.75(m,2H),6.72(d,J=8.2 Hz,1H),3.78(s,3H),3.17(s,3H),2.25(s,3H),2.24~2.03(m,3H),1.98~1.88(m,1H),1.69(brs,1H),1.34(s,9H).13C NMR(100 MHz,CDCl3)δ:180.27,156.22,136.82,135.22,111.99,110.25,108.36,55.91,47.72,47.62,38.21,36.34,26.37,24.40. FTIR(KBr,thin film,cm-1)ν:3 397,2 970,1 701,1495,1049.HRMS(ESI-TOF)m/z([M+H]+,C14H21N2O2)计算值249.159 8,实验值249.159 4.
2.3.9 (3aS,8aR)-5-甲氧基-1,3a,8-三甲基-1,2,3,3a,8,8a-六氢吡咯[2,3-b]吲哚(1)的合成 在双口圆底烧瓶中,称取化合物18(124 mg,0.5 mmol),溶解于5 mL无水四氢呋喃中,氮气保护下脱气3次,0℃冰水浴下,缓慢加入DIBAL-H(1.0 mol/L,1.5 mL,1.5 mmol),然后缓慢升至室温反应2 h,最后在80℃油浴加热下反应15 h.反应完后,将反应冷却至0℃,缓慢加入5 mL饱和酒石酸钾钠溶液,在室温剧烈搅拌2 h,加入水(10 mL)稀释,乙酸乙酯(15 mL×3)萃取,无水硫酸钠干燥,滤液减压浓缩,将浓缩后得到的粗产品,经柱层析纯化[V(石油醚)∶V(乙酸乙酯)=1∶2]得到淡黄色油状液体1(102 mg,0.44 mmol),收率88%.1H NMR(400 MHz,CDCl3)δ:6.70~6.61(m,2H),6.36(d,J=8.2 Hz,1H),4.04(s,1H),3.75(s,3H),2.89(s,3H),2.72(dt,J=8.8,5.4 Hz,1H),2.63(dt,J=8.9,7.4 Hz,1H),2.53(s,3H),1.94(dd,J=7.4,5.4 Hz,2H),1.43(s,3H).13C NMR(100 MHz,CDCl3)δ:153.07,146.74,138.43,112.28,109.95,107.59,98.50,56.16,53.34,52.88,40.98,38.34,38.12,27.59.FTIR(KBr,thin film,cm-1)ν:3 392,2954,2862,1 496,1 279,1 219,1120,1031.HRMS(ESI-TOF)m/z([M+H]+,C14H21N2O)计算值233.1649,实验值233.164 7.化合物的对映体过量(ee),在大赛璐(Daicel)Chiralpak OD-H柱上测定,溶剂为V(正己烷)/V(异丙醇)=98∶2,流速0.5 mL/min,检测波长254 nm,保留时间:t1=10.7 97 min [(S, R)-异构体],t2=13.4 70 min[(R,S)-异构体],对映体过量(ee)=63%,分析结果与已知报道的文献相一致[15]。
3 结论
综上所述,本文报道了一条基于铜催化邻溴苯胺类化合物芳基化串联烷基化关键策略,以较好产率和中等的非对映选择性,完成了天然产物Esermethole中C3a全碳季碳手性中心的构建,进而通过后续的一些化学转化,实现了六氢吡咯吲哚生物碱天然产物Esermethole的高效合成.该合成路线中大多数反应步骤能以克级或十几克级规模顺利进行,可以大量制备天然产物Esermethole及其类似物,为相关化合物的活性研究提供物质基础;同时该合成路线具有较好的普适性,此方法还可以实现一些含有季碳手性中心的六氢吡咯吲哚类生物碱天然产物合成,目前该项研究工作还在有序进行中,后续我们将继续报道该类天然产物的合成研究.
注:所有新化合物的1H NMR,13C NMR,DEPT135,DEPT90谱图以及化合物1手性HPLC的谱图等参见网络电子版附件支撑材料.
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
昆明医科大学学报(2020年12期)2021-01-26
山东化工(2019年11期)2019-06-26
吉林农业(2019年6期)2019-06-11
国际呼吸杂志(2019年1期)2019-03-08
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
合成化学(2015年10期)2016-01-17
华东师范大学学报(自然科学版)(2014年4期)2014-03-11
