
气相色谱-串联质谱法测定动植物源性食品中氯苯胺灵残留量
2021-06-04 02:17周若浩吕岱竹梁水连相坛坛王明月
食品科学 2021年10期
周若浩,吕岱竹,马 晨,梁水连,3,相坛坛,王明月,3,*
(1.华中农业大学食品科技学院,湖北 武汉 430070;2.中国热带农业科学院分析测试中心,海南 海口 571101;3.海南大学热带作物学院,海南 海口 570228)
氯苯胺灵是一种氨基甲酸酯类除草剂,其作用机理是抑制β-淀粉酶的活性、抑制植物RNA和蛋白质的合成、干扰氧化磷酸化和光合作用、破坏细胞分裂,常用于防除一年生禾本科杂草和少数一年生阔叶杂草[1]。氯苯胺灵除草效力强、持效期长、对作物本身损害小,在提高果蔬作物产量、增加产品效益方面发挥了重要作用[2]。但是氯苯胺灵在果蔬种植中的不当或过量使用易造成果蔬食品中农药高残留问题,并可随着食物链和水循环影响草食性动物以及人体健康[3-4]。欧盟的一项消费者风险评估指标表明,氯苯胺灵对消费者和非靶标节肢动物具有潜在的内分泌干扰特性,且具有严重的慢性风险,决定不再续展氯苯胺灵,但我国还未采取相应措施[5-9]。因此,建立一种动植物源食品中氯苯胺灵残留量的分析方法十分必要,可为完善氯苯胺灵的残留限量标准、评估氯苯胺灵的膳食摄入风险提供科学依据。
气相色谱法以及气相色谱-质谱法作为农药残留检测手段在食品安全检测方面发挥着越来越重要的作用[10-22]。目前氯苯胺灵的检测方法主要为反相高效液相色谱法[23-25]、气相色谱法[26]和气相色谱-质谱法[27]等,已有报道的检测样品主要为马铃薯、土壤和生活用水[28-34]等,尚鲜见其在动植物源食品中残留检测的相关报道。但现有的前处理技术实验步骤繁琐、费时费力,不适用于大批量样品的检测。本研究采用QuEChERS(quick, easy, cheap,effective, rugged, safe)及固相萃取法对动植物源食品中的氯苯胺灵进行提取净化,使用气相色谱-串联质谱(gas chromatography-tandem mass spectrometry,GC-MS/MS)进行分析,操作简便、回收率高、重复性好,适用于大批量样品检测,以期为动植物源食品中氯苯胺灵的残留检测提供依据。
1 材料与方法
1.1 材料与试剂
香蕉、牛肉、纯牛奶 海南海口市售。香蕉全果切段后用匀浆机匀浆至黏稠半固体状,牛肉样品用均质机绞碎至无明显肉块程度,将处理好好的样品置于-20 ℃冷冻保存,牛奶样品常温保存,待测。
99.5 %氯苯胺灵标准品 德国Dr. Ehrenstorfer GmbH公司;乙腈、正己烷、丙酮、甲醇(均为色谱纯)赛默飞世尔科技有限公司;分散固相萃取填料N-丙基乙二胺(primary secondary amine,PSA)、石墨化碳黑(graphitized carbon black,GCB)、C18,固相萃取C18柱、NH2柱和Florisil硅土柱 上海安谱实验科技股份有限公司;实验用水均为超纯水。
1.2 仪器与设备
TRACE 1300气相色谱仪、TSQ9000三重四极杆质谱仪 赛默飞世尔科技有限公司;Milli-Q Advantage A10超纯水系统 德国默克密理博公司;CR22N落地式高速冷冻离心机 日本日立公司;AL204电子天平 上海梅特勒-托利多仪器有限公司;KQ-500DE数控超声波清洗器 昆山市超声仪器有限公司;UMV-2多管旋涡混合器 北京优晟联合科技有限公司;R-210控温控压全自动旋转蒸发仪 上海豫康科教仪器厂。
1.3 方法
1.3.1 样品前处理
1.3.1.1 植物源样品
称取10 g匀浆好的样品于50 mL塑料离心管,加入20 mL正己烷和2 g氯化钠,以2 500 r/min涡旋6 min,以8 000 r/min离心5 min。取上清液5 mL,加入100 mg PSA净化,以2 500 r/min涡旋10 min,以8 000 r/min离心5 min,将所有溶液转移至50 mL圆底烧瓶中,于40 ℃水浴锅中旋转蒸发至干,用1 mL正己烷溶解,过0.22 μm滤膜,转入进样小瓶中待测。
1.3.1.2 动物源样品
称取10 g(牛奶取10 mL)匀浆好的样品于50 mL塑料离心管,加入20 mL正己烷和2 g氯化钠,以2 500 r/min涡旋6 min,以8 000 r/min离心5 min。取上清液5 mL,转移至已被活化的NH2固相萃取柱中,待净化完毕后用5 mL正己烷作为淋洗液洗柱,收集全部流出液于50 mL圆底烧瓶中,于40 ℃水浴锅中旋转蒸发至干,用1 mL正己烷溶解,过0.22 μm滤膜,转入进样小瓶中待测。
1.3.2 色谱条件
色谱柱:HP-5MS毛细管柱(0.25 mm×60 mm,0.25 μm);升温程序:初始温度100 ℃以10 ℃/min的速率升至200 ℃,保持1 min,以30 ℃/min的速率升至280 ℃,保持2 min;载气(He)流速1.0 mL/min,进样量1 μL;进样口温度:250 ℃:进样方式:无分流进样。
1.3.3 质谱条件
电子电离源,电离电压:70 V;离子源温度:300 ℃;传输线温度温度:280 ℃;定性离子m/z(碰撞能量):213(14 eV);定量离子:127(14 eV),171(6 eV)。
1.3.4 基质标准溶液的配制及标准曲线绘制
基质效应在农药残留分析中不可避免,不同农药在不同的基质中效应不同,可能会影响农药残留量的测定。本实验通过对溶剂曲线和基质曲线进行比较,发现以溶剂曲线作为定量标准时,氯苯胺灵在植物源性食品中的加标回收率在118.3%~127.4%范围内,在动物源性食品中的加标回收率在55.6%~71.3%范围内,这说明氯苯胺灵在植物源性食品基质中呈现基质增强效应,在动物源性食品中呈现基质减弱效应。以基质标准曲线进行定量时,氯苯胺灵的加标回收率在80.6%~107.8%范围内,满足农药残留检测要求,因此均需以基质标准曲线进行定量。
称取适量氯苯胺灵标准品,用正己烷定容至10 mL,得到1 000 μg/mL的氯苯胺灵标准储备液,用正己烷逐级稀释至质量浓度为1 μg/mL,再用空白基质提取液分别稀释至0.01、0.05、0.1、0.2、0.5 μg/mL,以氯苯胺灵质量浓度为横坐标,对应的质谱峰面积为纵坐标,绘制基质标准曲线。
2 结果与分析
2.1 仪器条件的优化
2.1.1 色谱条件的优化
氯苯胺灵为弱极性化合物,故选用弱极性的HP-5MS毛细管柱(0.25 mm×60 mm,0.25 μm)进行分离。色谱条件优化的主要目的是在获得良好化合物峰形的基础上尽量缩短分析时间,对柱子的升温程序进行选择,实验得出:初始温度100 ℃,以10 ℃/min速率升至200 ℃,保持1 min,以30 ℃/min的速率升至280 ℃,保持2 min条件能得到良好的氯苯胺灵峰形,为了提高检出限又要防止进样饱和,选择进样量为1 μL。
2.1.2 质谱条件的优化
采用单离子检测扫描(single ion monitoring,SIM)和选择反应监测(selective reaction monitoring,SRM)两种模式对氯苯胺灵进行检测分析。通过比较,发现采用SRM模式比采用SIM模式灵敏度更高、择性更好,抗干扰能力更强,更适合于复杂基质中农药残留的检测。在SRM模式下,进行质谱参数的优化。设定不同的碰撞能量对母离子进行轰击得到子离子二级质谱扫描图,从中选择响应值最高的2 个碎片离子作为子离子,改变碰撞电压优化各组母离子和子离子。最后选择响应最好的组合,确定氯苯胺灵SRM的最佳质谱条件,见表1。

表1 氯苯胺灵质谱条件Table 1 Mass spectrometric conditions for chlorpheniramine
2.2 前处理条件的优化
2.2.1 提取溶剂的优化
本研究通过比较不同提取溶剂(正己烷、丙酮、乙腈)下氯苯胺灵的回收率,以确定检测氯苯胺灵的最优提取溶剂,结果见图1。本研究考察不同提取溶剂对于动植物源食品中氯苯胺灵回收率的影响,发现采用丙酮进行提取时回收率在74%~81%之间,可能是由于氯苯胺灵在丙酮中溶解度较小。利用正己烷和乙腈作为提取溶剂时回收率分别在98%~102%之间及89%~97%之间,稳定性较好,均可作为提取溶剂,考虑到正己烷回收率更准确,因此选择正己烷作为提取溶剂。

图1 提取溶剂对氯苯胺灵回收率的影响Fig.1 Recoveries of chlorpheniramine using different extraction solvents
2.2.2 提取时间的优化
样品经溶剂提取,提取时间不同也会影响农药的回收率。本研究比较同一种溶剂不同提取时间下对氯苯胺灵回收率的影响,结果如图2所示。在分别加标量为0.1、0.2、0.3 μg/mL氯苯胺灵的情况下,在提取时间为2 min时,样品回收率偏低,表明样品中的农药未被充分提取。随着提取时间延长,氯苯胺灵的回收率先上升后趋于平缓,约在6 min达到最大回收率,因此选择提取时间为6 min。

图2 提取时间对氯苯胺灵回收率的影响Fig.2 Recoveries of chlorpheniramine at different extraction times
2.2.3 分散固相萃取填料的选择

图3 净化剂种类和用量对氯苯胺灵回收率的影响Fig.3 Recoveries of chlorpheniramine as a function of purifier dosage using different purifiers
各种分散固相萃取填料对样品的净化作用各不相同,本研究比较不同分散固相萃取填料净化对果蔬中氯苯胺灵回收率的影响。由图3可知,随着各种不同填料用量的增加,氯苯胺灵的回收率基本保持先增加后减少的规律。C18及GCB填料用量过大时,氯苯胺灵回收率较低,这可能是对目标物也产生了一定的吸附作用。因此选择PSA填料作为净化剂,经过对比发现PSA填料在100 mg用量时效果最佳,故选择100 mg PSA填料作为净化剂。
2.2.4 固相萃取柱的选择
固相萃取柱具有灵敏度高、重复性好、可批量处理等优点,比较C18柱、NH2柱和Florisil柱净化对动物食品中氯苯胺灵回收率的影响,结果如图4所示,C18柱对于氯苯胺灵的吸附能力过强,导致氯苯胺灵回收率偏低,在3 种固相萃取柱的净化下,氯苯胺灵回收率在85.8%~103.5%,NH2柱对氯苯胺灵净化效果好,回收率高,因此选用NH2柱进行净化。
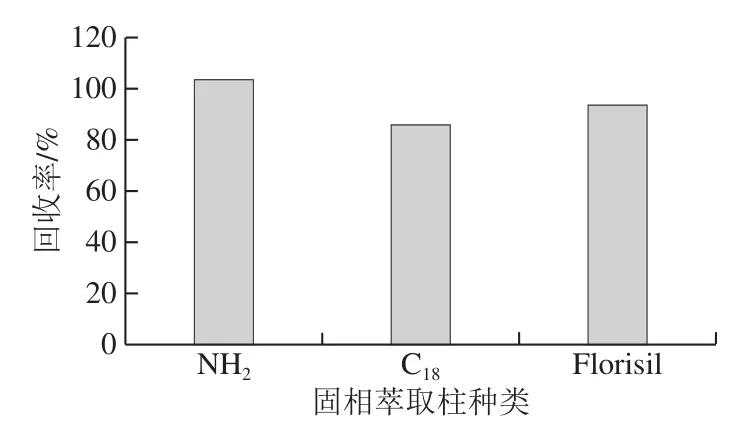
图4 固相萃取柱对氯苯胺灵回收率的影响Fig.4 Recoveries of chlorpheniramine using different SPE columns
2.3 线性范围、方法检出限及定量限
为了消除基质效应对定量结果的影响,实验采用空白样品提取液配制基质匹配标准溶液。在以上条件下,以氯苯胺灵质量浓度为横坐标(x),质谱峰面积(y)为纵坐标,线性关系为y=2.39×108x-1.885×106,R2=0.999 7,在0.01~0.5 μg/mL范围内的线性关系良好;以空白的牛肉基质提取液配制基质标准溶液,线性关系为y=1.29×105x-6.23×105,R2=0.999 3,在0.03~1 μg/mL范围内的线性关系良好;以空白的牛奶基质溶液配制基质标准溶液,线性关系为y=1.50×105x-1.50×105,R2=0.999 5,在0.03~1 μg/mL范围内的线性关系良好。分别以最低限加标样品的基线噪音值,当加标量为1.0 μg/kg时,氯苯胺灵特征峰峰高为27 491,基线噪音峰高为18 575,计算得出信噪比RSN=1.48,按照RSN=3计算得氯苯胺灵的检出限为2.0 μg/kg,按照RSN=10计算得氯苯胺灵定量限为6.7 μg/kg。
2.4 回收率与相对标准偏差(relative standard deviation,RSD)实验结果
称取不含目标物的动植物源食品样品,在植物源食品香蕉中分别添加0.02、0.04、0.4 mg/kg 3 个添加量的氯苯胺灵标准溶液,在动物源食品中分别添加0.01、0.02、0.1 mg/kg 3 个添加量水平的氯苯胺灵标准溶液,按照所建立的方法进行前处理以及GC-MS/MS测定。每份样品平行测定5 次,考察方法的回收率和精密度。3 种基质中方法回收率和RSD如表2所示,其在植物源食品香蕉中方法回收率为96.1%~105.7%,RSD为2.3%~6.5%;在动物源食品牛肉中方法回收率为80.6%~102.8%,RSD为5.7%~8.5%;在动物源食品牛奶中方法回收率在95.0%~107.8%,RSD为7.5%~8.4%,可见该方法具有良好的可靠性。

表2 动植物源食品中氯苯胺灵的回收率和精密度(n=5)Table 2 Recoveries and precision of chlorpheniramine from actual food samples (n= 5)
2.5 实际样品检测结果
从海南海口本地农贸市场分别随机抽取20 份动植物源食品样品,对其进行氯苯胺灵残留检测。在一份植物源样品中检出氯苯胺灵,含量为19.4 μg/kg;在一份动物源性样品中检出氯苯胺灵,含量为12.6 μg/kg,其余样品均未检出氯苯胺灵。
3 结 论
本研究开发动植物源食品中氯苯胺灵的检测分析方法,以动植物源食品基质提取液配制的氯苯胺灵标准溶液作为定量标准,减少了基质效应对实验结果的影响,基质标准溶液在检测范围内线性关系良好,相关系数高于0.999。同时优化前处理过程中的提取和净化方法,回收率在80.6%~107.8%之间,RSD为2.3%~8.5%,操作简便、重复性好,适用于批量样品检测,可很好满足动植物源食品中氯苯胺灵残留的检测分析,为完善氯苯胺灵的残留限量标准、评估氯苯胺灵的膳食摄入风险提供了科学依据。
猜你喜欢
能源化工(2022年1期)2023-01-14
化工设计(2022年4期)2023-01-02
含能材料(2022年10期)2022-10-22
现代仪器与医疗(2022年1期)2022-04-19
石油炼制与化工(2022年2期)2022-02-15
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
食品安全导刊(2021年20期)2021-08-30
现代仪器与医疗(2021年2期)2021-07-21
化工管理(2020年26期)2020-10-09
