
细菌核心基因组多位点序列分型(cgMLST)与溯源评价
2021-05-28 02:41朱丽萍张文成颜世敢陈蕾蕾崔生辉
畜牧与兽医 2021年6期
朱丽萍,张文成,颜世敢*,陈蕾蕾,崔生辉
(1. 齐鲁工业大学生物工程学院/山东省微生物工程重点实验室,山东 济南 250353;2. 山东省农业科学院农产品研究所,山东 济南 250100;3. 中国食品药品检定研究院,北京 100050)
食品安全关系人民健康和国计民生,是全球关注的热点问题。食源性疾病是引发食品安全事件的主要因素。全球每年约15亿人患食源性疾病,其中约70%是因食品被微生物污染所致。我国46.4%的食品安全事件是由食源性致病菌引起。沙门菌、致病性大肠杆菌、李氏杆菌等是最重要的食源性致病菌。其中沙门菌引起的食物中毒在微生物性食物中毒中占比高达70%~80%,每年造成全球约1.15亿人感染和37万人死亡[1]。加强对沙门菌等食源性致病菌的监控和防控,意义重大且迫在眉睫。
细菌分型是细菌溯源、流行病学调查、食品安全监管的重要手段,对食品安全、公共卫生具有决定性意义。
1 细菌溯源分型技术概述
细菌溯源分型技术包括表型分型技术和基因分型技术两类。表型分型技术有血清分型、噬菌体分型等;基因分型技术有脉冲场凝胶电泳(pulsed field gel electrophoresis,PFGE)、多位点序列分型(multilocus sequence typing,MLST)、核糖体多位点序列分型(ribosome multilocus sequence typing,rMLST)、全基因组单核苷酸多态性(whole genome single nucleotide polymorphism,wgSNP)、全基因组多位点序列分型(whole genome multilocus sequence typing,wgMLST)、核心基因组多位点序列分型(core genome multilocus sequence typing,cgMLST)等。目前常用的细菌溯源分型技术以血清学分型、PFGE、MLST为主。血清学分型依靠肉眼观察血清型试验是否出现凝集而判断细菌的抗原型,试验误差大,分辨率低,再加上细菌的血清型众多,如沙门菌有2 610种血清型[2],血清学分型的工作量大、耗时长,分型效果不理想。PFGE是根据基因组酶切片段的电泳条带图谱进行聚类分析,分辨率及重复性较好,但无法辨别酶切位点之外的基因序列变异,且分型结果受人为因素影响大[3-4]。MLST是基于7~9个管家基因的序列多态性建立的分型技术,重复性、可比性好,但分辨率仍不能满足精准溯源的需求,无法反映基因组其他几千个基因的差异[5]。
随着基因组测序技术的普及和测序成本的降低,基于全基因组序列的分型技术迅速发展成为细菌溯源的主流技术[6]。全基因组分型技术以wgMLST、cgMLST、wgSNP等为代表,分辨率高,重复性好,能够实现精准溯源[7]。但wgMLST、wgSNP分析消耗巨大的计算资源,一般实验室难以实现准确分型[8],而且wgSNP对测序的准确性要求极高,必须确保SNP位点的准确性和一致性。cgMLST与wgMLST、wgSNP相比,分辨率相当,仅需要较少的计算能力,可操作性和应用性更强。
cgMLST已广泛用于沙门菌[1,5,7-9]、耶尔森菌[5]、大肠杆菌[10-11]、李氏杆菌[12-15]、布氏杆菌[16-17]、金黄色葡萄球菌[18]、猪链球菌[19]、铜绿假单胞菌[20]、克雷伯菌[21]、结核杆菌[22-23]、鸡支原体[24-26]、弯曲杆菌[27-28]等危害人、畜禽的致病菌基因组分型中。由于缺乏统一的分型标准,不同研究者采用的分型方案不同,导致分型结果缺乏可比性。目前为止,尚未见细菌全基因组溯源分型指南的报道。本文通过制定细菌的cgMLST指南,来规范cgMLST分型操作,使cgMLST分型结果具有可比性、重复性,便于分型结果的共享与比较。
2 细菌cgMLST分型与溯源的原理
cgMLST是基于细菌的核心基因组进行的多位点序列分型技术[29],分型精度高,重复性、可比性好,能客观揭示细菌的遗传进化关系,为细菌溯源、流行病学调查提供了科学依据[30]。
cgMLST是MLST的升级。MLST是建立在7~9个管家基因的基础上的,无法反映其他大量基因的异同;而cgMLST是建立大量保守基因基础上的,具有更高的分辨率,分型结果更客观、更精准。而且用于cgMLST分型的核心基因中包含了用于MLST分型的所有管家基因,这样便于将cgMLST结果与已有的MLST分型结果进行比较,检验cgMLST分型结果的可靠性。
cgMLST具有高分辨率,能将一种细菌分为多个亚型,通过聚类分析确定不同亚型间的遗传进化关系,实现更深层次的溯源。cgMLST逐渐成为细菌分型、溯源、分子流行病学研究的重要手段,分型结果可用于不同实验室间的共享和比较[31]。
2.1 核心基因组的特征
细菌的核心基因是指某种细菌的不同菌株共有的一批保守基因,它们负责该种菌的生物学基本特征及主要表型特征。核心基因具备以下特点:单拷贝基因,即在基因组中只出现一次;非质粒基因;基因内没有无效的起始/终止密码子;与其他基因不重叠(overlap);等位基因间不同源。
核心基因组是指根据分型需要人为地将某种细菌不同菌株共有的一定数量的核心基因组合在一起形成的基因群。基于细菌核心基因组序列多态性的分型技术就是cgMLST。
2.2 核心基因组的确定方案
细菌核心基因组的确定有两种方案:根据菌株的基因组序列比对结果和分析目的,自行确定核心基因组中使用的核心基因的种类和数量多少。采用的核心基因不同,同一株菌获得的cgMLST分型结果不同,对溯源分析的精度有一定影响;采用EnteroBase、PubMLST、SISTR等分型数据库公开的核心基因名录[9]。其中EnteroBase数据库的数据量大、分辨率高、认可度最高[5]。
2.2.1 自行筛选核心基因组
选用已公开的同种细菌的完整基因组作为“种子”基因组(seed genome)。“种子”基因组需满足以下条件:基因组完整,最好是采用Sanger法测序,且已完成基因注释;基因组来自细菌的纯培养物;代表株的基因组。
可利用Ridom Seqsphere+、Bionumerics、BPGA(细菌泛基因组分析工具)等生物信息学软件从全基因组中筛选核心基因[32]。如 Ridom Seqsphere+分型软件内嵌模块自动把种子基因组内满足条件的等位基因全部筛选出来作为核心基因组。
2.2.2 EnteroBase数据库中公开的核心基因组
EnteroBase数据库中公开了沙门菌(Salmonella)、大肠杆菌/志贺菌(Escherichia/Shigella)、艰难梭菌(Clostridioides)、弧菌(Vibrio)、螺杆菌(Helicobacter)、耶尔森菌(Yersinia)、莫拉菌(Moraxella)共8种细菌的核心基因组,核心基因的名称及功能见EnteroBase在线分析网站(http://enterobase.warwick.ac.uk/species/)。其中沙门菌的cgMLST分型方案中包含3 002个等位基因[33]。EnteroBase数据库中用于cgMLST分型的核心基因的种类是固定的,用户无法改变核心基因的种类和数量,也无法改变分型使用的算法。通过这种固定化的cgMLST分型方案,可以做到不同分析者采用EnteroBase数据库对同一株菌进行cgMLST分型获得的结果具有一致性。
2.3 cgMLST型的赋值规则
通过BLAST、Usearch等分析工具,检索各分离株基因组序列中的基因位点,与分析模板中的核心基因组的序列进行比对,匹配度≥70%的基因序列被归类为等位基因,经后续分析后将对其进行赋值;匹配度<70%的基因序列则不再进行后续分析,该等位基因的基因型标记为缺失(Missing)或赋值-1。
所有与模板基因组中的核心基因序列的匹配度≥70%的等位基因被分别赋值,每个等位基因被单独赋予一个正整数,不同的数值代表不同的基因型,数值之间不存在关联性,数值大小表示提交时间的先后顺序。事先定义各等位基因使用的模板基因组的核心基因的基因型都为“1”,待分析的等位基因序列与模板基因组的核心基因进行BLAST比对,序列相同的等位基因则基因型赋值也为“1”,序列不同的等位基因则按照提交时间的先后顺序赋予一个新的基因型数值。同一个数据库内的等位基因编号与基因序列严格一一对应,后续用于分析的基因序列与已经赋值的基因型所对应的序列相同时,重复使用该基因型编号。不同的数据库因为采用的核心基因组不同,所赋予的同株菌的基因型编号不同。
每株菌的等位基因型编号按照指定的顺序排列形成该菌的等位基因谱。
cgMLST型的赋值:为了与MLST分型的ST型区分,cgMLST型简写为cgST。赋予种子基因组的cgST型为1。每株菌的等位基因谱与数据库中已有的等位基因谱比对,如果相同则被赋予相同数值的cgST型;如果不同,则按照向数据库提交时间的先后顺序顺位编号赋予一个新的正整数,作为该基因组的cgST基因型。每种cgST型对应唯一的等位基因谱。
2.4 基于cgMLST的细菌基因组溯源分析
cgMLST分型后往往要通过绘制遗传进化树,分析菌株间的遗传距离和亲缘关系,实现细菌溯源分析。
绘制进化树可采用Ridom、Bionumerics或EnteroBase在线分析平台。在完成cgMLST分型后直接绘制遗传进化树;也可以将进化树数据导出为.nwk文件,利用进化树修饰软件(如Figtree、Meqsuite、Dendroscope)进一步修饰。
进化树的种类有邻居加入树(Neighbor Join Tree,NJT)、最小生成树(Minimum Spanning Tree,MST)、葡萄树(GrapeTree)等[34]。不同类型的进化树的形状不同,但都可用于细菌溯源分析。绘制邻居加入树、葡萄树采用的算法是N-J算法;绘制最小生成树采用的算法有Kruskal算法或Prim算法。溯源分析时MST的呈现形式更直观。
EnteroBase在线分析平台还提供了基于cgMLST的HierCC层次聚类法,可以不用绘制进化树仅依靠HierCC型数值大致判断2株菌间的核心基因差异的个数。如果2个细菌的3 002个核心基因中存在2个等位基因差异,2株菌的cgMLST分型不同,即HierCC0聚类时为不同类,但HierCC2聚类时为同一类,且HierCC5、10….聚类时均为同一类。
cgMLST分型后,对于等位(核心)基因差异数≤10的2株菌,判定其具有高度同源性;等位(核心)基因差异数在10~30个时,需要结合流行病学调查与菌株背景信息判断菌株间的同源性。
3 细菌cgMLST分型使用的数据和生物信息学软件
3.1 细菌cgMLST分型采用的基因组数据类型
细菌cgMLST分型是建立在基因组高通量测序序列的基础上进行的。细菌基因组DNA在二代测序仪上测序,得到原始数据(Raw Data),过滤掉低质量的reads,获得有效数据(Clean Data)。使用组装、拼接、优化、质控软件对Clean Data进行组装、拼接、优化和补洞,过滤掉500 bp以下的片段,最终得到基因组序列(Sequence)。
Raw Data、Clean Data、Sequence都可用于细菌cgMLST分型。但不同的基因组分型软件采用的基因数据类型有差别。
下载的细菌基因组序列:可以从基因组数据库(如GenBank)中下载已发布的细菌基因组序列,用作cgMLST分型的基因组数据。
3.2 细菌cgMLST分型需要的生物信息学软件
3.2.1 Bionumerics(Version 7.6,Applied-Maths, Belgium)
适用数据类型:二代测序Clean Data、拼接的基因组序列及基因组数据库中的基因组序列文件。Bionumerics的wgMLST分型方案不能自行创立。沙门菌的全基因组分型是基于21 065个等位基因[13]。Bionumerics的wgMLST分型数据库是Applied-Maths公司自己创建的云数据库,使用者无法改变分型方案,不同分析者获得的分型结果一致。
优点:本地软件加在线数据库分析,对计算资源配置要求高,Bionumerics软件的分析功能强大,主要用于细菌wgMLST分析,向下兼容MLST、cgMLST分型,提供多达42种常见微生物的全基因组分型模板。
缺点:软件使用费较高。
3.2.2 Ridom Seqsphere+(Version 5.1.0,Ridom GmbH,Germany)
适用数据类型:二代测序Clean Data、拼接的基因组序列及基因组数据库中的基因组序列文件。cgMLST分型方案采用的核心基因组是EnteroBase提供的cgMLST分型方案或通过基因组比对自行创立分型方案。通过本地化软件分析,结合软件分型命名数据库cgMLST.org,实现cgMLST分型。
优点:本地化分析,能够对分型结果进行多种进化树绘制,操作较简单,用户可自行创立分型方案进行个性化分析。
缺点:软件使用费高,占用大量的本地计算资源,对计算机硬件要求高。
3.2.3 EnteroBase免费在线分析平台(http://enterobase.warwick.ac.uk/)
该分析平台免费注册使用,但目前仅能进行沙门菌(Salmonella)、大肠杆菌/志贺菌(Escherichia/Shigella)、艰难梭菌(Clostridioides)、弧菌(Vibrio)、螺杆菌(Helicobacter)、耶尔森菌(Yersinia)、莫拉菌(Moraxella)等8种肠杆菌的cgMLST分型和溯源。其中Salmonella分型数据库包含262 196株菌,有MLST和cgMLST分析模块;Escherichia/Shigella分型数据库包含147 636株菌,有MLST和cgMLST分析模块;Clostridioides分型数据库包含18 371株菌,有MLST、cgMLST、rMLST分析模块;Vibrio分型数据库包含11 364株菌,有rMLST分析模块;Helicobacter分型数据库包含5 477株菌,有rMLST分析模块;Yersinia分型数据库包含4 915株菌,有MLST和cgMLST分析模块;Moraxella分型数据库包含2 564株菌,有MLST和rMLST分析模块(以上数据截至2020-07-16)。
适用数据类型:二代测序Clean Data,或高质量的完整基因组序列(自己拼接的基因组序列和下载的基因组序列)。cgMLST V2+分型方案的特点:在线分析平台,权威性高,使用广。
优点:分析全部通过在线服务器实现,占用的计算机资源较低,对计算机配置要求不高。固定化的分型程序能够保证分型参数一致,最大程度保证分型结果的可比性。用户只需上载基因数据便可得到分型结果。在基因组质量要求,EnteroBase仅接受由平台自身通道生成的组装基因组且符合重叠群大小N50≥20 kb,基因组大小≥4 Mb并且至少含有97%的核心基因。在数据方面,EnteroBase具有大量的可参比结果,能够调用更多的菌株进行遗传进化分析、聚类分析。
缺点:不能进行个性化设置,可操作程度低。上载数据只能是测序的Raw Data,不接受拼接后的基因组序列。
4 cgMLST分型的技术路线
cgMLST分型技术路线见图1。
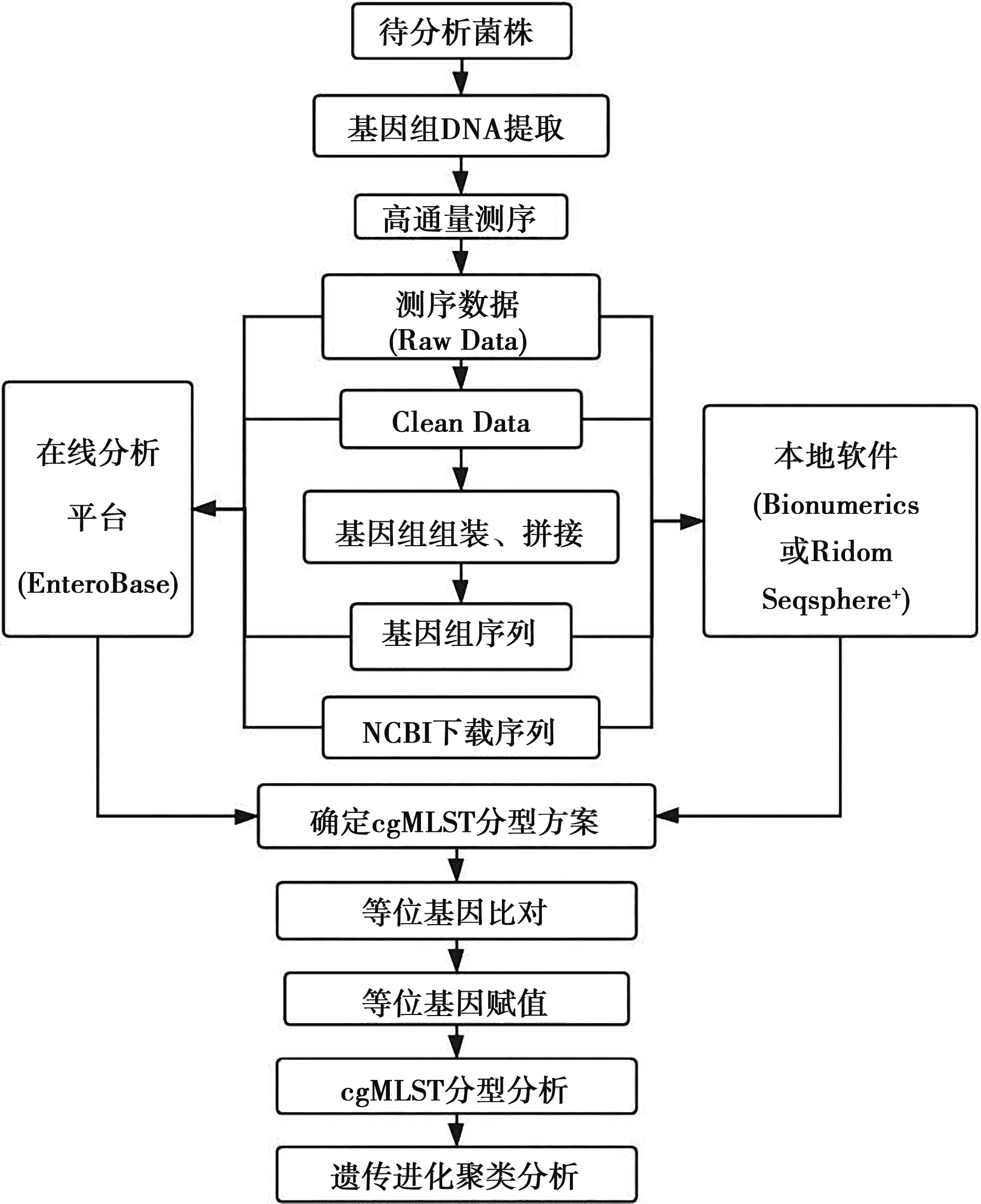
图1 细菌cgMLST分型技术路线
5 细菌cgMLST分型、溯源分析步骤
5.1 采用公开的cgMLST分型方案
5.1.1 使用Ridom Seqsphere软件自带的cgMLST分型方案
创建分析项目。打开Ridom Seqsphere软件,先登录服务器(https://www.ridom.de/seqsphere/)上的Ridom SeqSphere+,再启动客户端的Ridom SeqSphere+。点击菜单栏“File”,选择 “New” 里面的“Create Project”,进入分析项目的编辑界面,给待分析的项目命名及选择物种信息。本文以沙门菌为例,选择Salmonellaenterica。然后点击Download & Add,进入分型方案选择界面,从分型方案中单独勾选cgMLST或同时勾选MLST。Ridom SeqSphere+的沙门菌的cgMLST分型方案与EnteroBase中的沙门菌cgMLST v2方案完全相同。每个用于分析的基因组包含的等位基因与该等位基因参照序列进行BLAST,比对分析后保留同源性≥80%的结果,并给该等位基因进行基因型编号;若同源性<80%则该等位基因不会被编号,等位基因的基因型以及菌株的最终基因型命名来源于cgMLST.org数据库。点击“OK”完成项目创建。
细菌基因组的cgMLST分析。点击主界面的菜单栏 “Flie” 中的“Process Assembled Genome Data”,调出已创建的分析项目。添加基因组文件。添加本地基因组通过“Add from File”实现。软件也支持从NCBI在线获取基因组,但在线利用NCBI基因组进行分型分析耗时远大于本地基因组,所以建议先将基因组文件下载到本地磁盘再进行分析。选择待分析的基因组后,点击“OK”按钮,进行分析。若同时选择MLST与cgMLST两个模块,能同时获得两种分型结果。
分型结果的呈现与聚类分析。输入的基因组数据经cgMLST分析后,软件会自动弹出分析结果预览表。关闭预览表后,可在主界面点击“Tools”中的“Comparison Table”查看cgMLST分型结果。分析后的数据存储在每个项目下,可再次添加基因组分析数据,若同时选择MLST与cgMLST分析,会在表格下方出现MLST与cgMLST两个选择项目,选择其中之一便可获得该分析的全部结果。点击表格下方的Create Comparison Table便可进入分型结果。
分型结果以表格形式呈现。分型表从左到右各列分别为Missing Values in Distance Columns(缺失基因或因比对结果<80%造成该基因分型失败的个数)、Perc Good Targets(核心基因中被成功分型的等位基因的百分比,但只有等位基因携带率≥90%的样本才能用于新基因型的提交)、ST(MLST型,相同的ST型被标记成同一种颜色)、Complex Type(cgMLST型)。
点击分型结果界面上方菜单栏中的进化树分析按钮进行进化树的聚类分析,实现细菌的溯源分析结果可视化查看。
新产生的Complex Type的提交。cgMLST分析后,可能在Complex Type列中产生“?”特征值,这说明该基因组的Complex Type不存在数据库中,是一个新发现的细菌基因组,需要提交至数据库中获取一个新的Complex Type编号(即cgST型)。提交时,选中要提交的样本,点击右键,选择“Open Sample”,会在主界面形成一个上传表,完善上传表中的基因背景信息,包括分离人姓名、细菌分离时间、宿主来源等信息,填写完毕,点击上传按钮上传新产生的cgST型。
5.1.2 使用EnteroBase在线分析平台进行cgMLST分析
EnteroBase分析的数据对象是Clean Data,或高质量的完整基因组序列。
登录EnteroBase在线分析平台的网站(http://enterobase.warwick.ac.uk/),注册个人账户。
选择与待分析物种相对应的物种首页,进入Database Home。EnteroBase数据库中用于分析的物种模块有Salmonella、Escherichia/Shigella、Clostridioides、Vibrio、Helicobacter、Yersinia、Moraxella。
点击右侧Upload Reads进行基因组数据上载,填写相关信息,其中红色框为必填项,然后进行上载基因组数据。首次加载页面时,将显示一个空白行,在其中输入与上传菌株相关的数据。单击各单元格,可以通过直接键入或从下拉框中选择来输入数据,通过右键单击表格并选择“插入行” 来添加额外的行。
单击Read Files单元,出现一个对话框用于添加基因组文件。上载Illumina测序平台的原始数据文件(.fq.gz)。填写所有必填项后,“提交数据(Submit Data)”按钮将变为活动状态,点击进行上载。
查看基因组组装结果:返回物种首页,点击My Strains,进入个人数据界面。默认会进入基因组组装结果展示界面,没有标红的行即为组装成功,可进行后续分析。基因组组装结果的各项参数以表格形式展示,点击Status列的下载图标可下载组装基因组的序列文件。
cgMLST分型结果展示:通过点击右上角选择栏中的cgMLST V2 +HierCC V1查看cgMLST分型结果。cgMLST的基因型包括ST(cgST型)和不同HC数值(如HC0、HC2、HC5…HC2850)下的ST型。不同HC数值下的ST型之间不具有可比性。
Grapetree进化树绘制:点击GrapeTree图标,选择进化树算法(如N-J算法),弹出窗口完成Grapetree的绘制。可通过左侧的相关设定修饰进化树,最后导出分析结果,可以导出Newick Tree文件格式或者SVG图片格式。
5.2 自行创立分型方案
该方案适用于大多数细菌cgMLST分型,尤其是那些没有分型数据库的细菌的cgMLST分型。以利用Ridom软件为例,介绍如何自行创立分型方案进行沙门菌cgMLST分型。
登录软件:选择工具栏中的Tools下的cgMLST Target Definer,选择Seed Genome(可从NCBI上选择种子基因组序列)。选择添加或不添加“查询”基因组(Query Genomes)。添加质粒序列信息(该步骤可以使得与添加的质粒序列中的基因匹配度≥90%且长度>100 bp的基因不在核心基因列表中)。分析参数一般选择默认,创立分析模板。
打开创立的分析模板:导入待分析的基因组数据(组装的基因组序列或NCBI中的基因组文件),然后进行cgMLST分析。分析完成后,从任务栏创建分析表,补全菌株相关信息,勾选cgMLST分析项目,查看以图表形式展示的分型结果。
6 cgMLST分型与溯源分析的注意事项
种子基因组(或模板基因组):种子基因组的选择对cgMLST分型结果影响大。自行创立分型方案时选用的种子基因组(或模板基因组)要具有代表性、广泛性、完整性,尽量选用基因数据库中收录的完整基因组,不使用自行组装的分离株的组装基因组。
cgMLST分型方案中的核心基因组在不同菌株中要广泛分布且具有完整性。如沙门菌3 002位点分型方案满足在3 144个有代表性的沙门菌基因组中,每个等位基因至少存在于98%的基因组中,编码框架至少在每个基因组中的完整性≥94%。
分型参数的设置:可以使用软件的默认值,以保证获得的分型结果具有一致性。在同一数据库下,不同使用者按照相同的步骤、相同的参数,不同分析人员对同一基因组序列分析获得的分型结果基本一致,具有可比性。
物种匹配与否:提交的基因组序列要与选用模板基因组属于同一物种。
组装基因组的质量评价:衡量基因组组装质量的参数有Coverage、N50、Length、Contig Number、Low Quality等。通常要求基因测序覆盖倍数>100。选择合适的基因组重叠群参数,如二代测序的沙门菌组装基因组的重叠群大小需满足N50≥20 kb。原则上组装的基因组大小要大于已知该物种的最小基因组(如沙门菌基因组需>4.0 Mb,不排除基因组较小的特殊性)。
用于基因组测序的细菌必须为克隆株,不存在其他分离株的污染。提取的基因组DNA中也不存在其他物种DNA的污染。
7 小结
细菌溯源分型对食品安全监管、流行病学调查具有重要意义。目前常用的血清学分型、PFGE、MLST等细菌溯源分型方法存在分型精度低、工作量大、耗时长等缺陷,不能满足细菌精准溯源的需求。以wgMLST、cgMLST、wgSNP等为代表的全基因组分型技术具有分辨率高、重复性好的优点,能够实现细菌的精准溯源,逐渐成为细菌分型和溯源的主流技术。其中,cgMLST需要较少的计算能力,在细菌分型、菌株演变和暴发溯源等研究中将更具应用价值和发展潜力。
猜你喜欢
今日农业(2021年11期)2021-08-13
智慧健康(2021年17期)2021-07-30
国际检验医学杂志(2021年7期)2021-04-15
基层中医药(2020年5期)2020-09-11
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
基层中医药(2018年5期)2018-08-31
现代检验医学杂志(2016年5期)2016-08-20
制造技术与机床(2015年10期)2015-04-09
中国中医药现代远程教育(2014年21期)2014-03-01
遗传(2014年3期)2014-02-28
