
4,5-二取代苯并(氧化)呋咱类衍生物的合成及体外抗肿瘤活性
2021-05-27 07:31王晓慧王慧云谭文娟赵风兰杜广营孟庆国
烟台大学学报(自然科学与工程版) 2021年2期
王晓慧,王慧云,谭文娟,赵风兰,杜广营,孟庆国
(1.烟台大学新型制剂与生物技术药物研究山东省高校协同创新中心、分子药理和药物评价教育部重点实验室(烟台大学),山东 烟台 264005;2.济宁医学院药学院,山东 日照 276826)
某些含氮杂环化合物的结构与嘌呤、嘧啶等碱基的结构类似,具有较好的生物体内环境相容性,在新药研发方面有广泛的应用前景。苯并呋咱及其N-氧化物是有机化学中合成杂环的重要中间体,具有抗菌、抗寄生虫、舒张血管、抗肿瘤等广泛生物活性[1-2],其抗肿瘤机制可能与抑制肿瘤细胞核苷的磷酸化,限制核酸和蛋白质的合成,使DNA断裂有关[3]。且该类化合物是NO供体,在体内经巯基类化合物的作用可以缓慢释放氮氧化物,从而抑制肿瘤细胞生长和逆转肿瘤的耐药性[4-6]。
2011年,王洪波等[7]通过高通量筛选,发现4-硝基-7-(4-甲基哌嗪基)-苯并氧化呋咱(图1,XI-006)是一类能够抑制肿瘤细胞MDMX的表达,激活p53,保持肿瘤抑制基因活性的潜在抗肿瘤的代表化合物。本课题组前期研究发现,苯环上胺基和硝基的位置影响苯并(氧化)呋咱衍生物的体外抗肿瘤活性[8]。为进一步增强该类化合物抗肿瘤活性,获得结构新颖的化合物,以XI-006为先导化合物,设计、合成了10个4,5-二取代苯并(氧化)呋咱类新衍生物(图2)。并采用磺酰罗丹明B(SRB)比色法进行了体外抗肿瘤活性评价,发现化合物4a—4e对人非小细胞肺癌细胞NCI H460、乳腺癌细胞MCF-7及结肠癌细胞HCT-116具有较强的抑制作用,对MCF-7的IC50值小于1.0 μmol/L,对HCT-116的IC50值小于5.0 μmol/L,优于先导化合物XI-006。
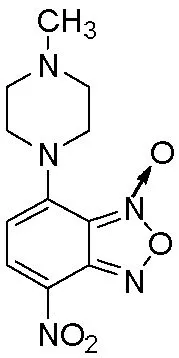
图1 XI-006的结构

图2 目标化合物4a—4e及7a—7e的合成路线
1 实验部分
1.1 仪器和试剂
WRS-1S数字熔点仪(上海易测仪器设备有限公司),温度未经校正;AVANCE-400型核磁共振仪(瑞士Bruker公司);Mariner型质谱仪(美国Applied Biosystems公司);薄层层析硅胶(烟台市化学工业研究所);试剂均为市售分析纯。
1.2 合成步骤
1.2.1 2-硝基-4-氯叠氮苯(1)的合成 250 mL反应瓶,加入30.0 mL乙酸、2-硝基-4-氯苯胺(2.75 g, 15.94 mmol)和10.0 mL浓硫酸,搅拌至全溶,降至0 ℃以下,缓慢滴加50.0 mL亚硝酸钠(1.15 g, 16.67 mmol)水溶液,5 h反应完全。滴加30.0 mL叠氮化钠(1.05 g, 16.15 mmol)水溶液,转移至室温反应,TLC检测至反应不再正向进行,反应体系用饱和氢氧化钠溶液调pH值至7,用适量乙酸乙酯溶解,有机相依次用水和饱和氯化钠溶液洗涤,无水硫酸钠干燥。过滤,减压浓缩,得黄褐色固体1(2.92 g),收率92.3%,直接用于下步反应。
1.2.2 5-氯-苯并氧化呋咱(2)的合成 按文献[9]方法合成,得亮黄色固体2(2.13 g),收率85.1%。直接用于下步反应。
1.2.3 4-硝基-5-氯-苯并氧化呋咱(3)的合成 100 mL反应瓶,加入12.0 mL浓硫酸和化合物2(1.02 g, 6.0 mmol),搅拌至全溶,降至0 ℃以下,缓慢滴加发烟硝酸(0.54 g, 8.57 mmol)和5.80 g浓硫酸混合液,4 h反应完全。加入适量乙酸乙酯,有机相依次用水和饱和氯化钠溶液洗涤,无水硫酸钠干燥。过滤,减压浓缩,硅胶柱层析,得黄色固体3(1.06 g),收率82.0%,mp:138~140 ℃。直接用于下步反应。
1.2.4 5-氯苯并呋咱(5)的合成 按文献[10]方法合成,得褐色油状物5(1.84 g),收率98.9%。直接用于下步反应。
1.2.5 4-硝基-5-氯苯并呋咱(6)的合成 100 mL反应瓶,加入11.0 mL浓硫酸和5(0.94 g, 6.0 mmol),搅拌至全溶,降至0 ℃以下,缓慢滴加发烟硝酸(0.51 g, 8.09 mmol)和6.1 g浓硫酸混合液,4 h反应完全。将反应液倒入碎冰中,抽滤,用水洗涤滤饼,室温干燥,得棕色固体6(446 mg),收率36.7%。直接用于下步反应。
1.2.6 4-硝基-5-(4-甲基哌嗪)-苯并氧化呋咱(4a)的合成 50 mL反应瓶,加入20.0 mL无水乙醇、3(100 mg, 0.46 mmol)和三乙胺(0.1 mL, 0.65 mmol),缓慢滴加5 mL 1-甲基哌嗪(93 mg, 0.93 mmol)的无水乙醇溶液,室温反应14 h,反应完全。减压浓缩,剩余物用适量氯仿溶解,有机相依次用水和饱和氯化钠溶液洗涤,无水硫酸钠干燥。过滤,减压浓缩,硅胶柱层析,得红色固体4a(70 mg),收率34.8%,mp: 136~139 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.32(1H, d,J=8.56 Hz, 7-H-Ar), 6.24(1H, d,J=8.6 Hz, 6-H-Ar), 3.45(4H, t,J=4.24 Hz, piperazine-2, 6-H), 2.67(4H, s, piperazine-3, 5-H), 2.40(3H, s,-CH3)。ESI-MS,m/z: 280.0[M+H+]。参照化合物4a的合成方法,合成化合物4b—4e,其外观、总收率、熔点、1H NMR和ESI-MS 数据如下:
4-硝基-5-己胺基-苯并氧化呋咱(4b):橘红色粉末,收率40.7%,mp: 101~103 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.66(1H, s,-NH-), 8.34(1H, d,J=9.12 Hz, 7-H-Ar), 6.24(1H, d,J=9.16 Hz, 6-H-Ar), 3.46(2H, s,-CH2CH2CH2CH2CH2CH3), 1.29~1.35(8H, m,-CH2CH2CH2CH2CH2CH3), 0.87(3H, t,J=6.08 Hz,-CH3)。ESI-MS,m/z: 281.1[M+H+]。
4-硝基-5-(4-异丙基哌嗪基)-苯并氧化呋咱(4c):深红色粉末,收率32.9%,mp: 102~105 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.39(1H, d,J=8.8 Hz, 7-H-Ar), 6.46(1H, d,J=8.84 Hz, 6-H-Ar), 3.45(4H, t,J=4.44 Hz, piperazine-2, 6-H), 2.62(4H, t,J=4.68 Hz, piperazine-3, 5-H), 2.69~2.76(1H, m,-CH-), 1.0(6H, d,J=6.52 Hz,-(CH3)2)。ESI-MS,m/z: 308.1[M+H+]。
4-硝基-5-(4-异丁基哌嗪基)-苯并氧化呋咱(4d):棕色粉末,收率35.8%,mp: 135~138 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.40(1H, d,J=8.8 Hz, 7-H-Ar), 6.48(1H, d,J=8.84 Hz, 6-H-Ar), 3.46(4H, s, piperazine-2, 6-H), 2.50(4H, s, piperazine-3, 5-H), 2.10(2H, d,J=7.36 Hz,-CH2-), 1.75~1.85(1H, m,-CH-), 0.87(6H, d,J=6.48 Hz,-(CH3)2)。ESI-MS,m/z: 322.1[M+H+]。
4-硝基-5-高哌嗪基-苯并氧化呋咱(4e):棕色粉末,收率40.2%,mp: 95~97 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.10(1H, d,J=9.12 Hz, 7-H-Ar), 6.32(1H, d,J=9.16 Hz, 6-H-Ar), 4.10(2H, s, homopiperazine-2-H), 3.97(2H, s, homopiperazine-7-H), 2.76(2H, t,J=4.72 Hz, homopiperazine-3-H), 2.56(2H, t,J=5.2 Hz, homopiperazine-5-H), 2.28(3H, s,-CH3), 1.98~2.01(2H, m, homopiperazine-6-H)。ESI-MS,m/z: 294.1[M+H+]。
1.2.7 4-硝基-5-(4-甲基哌嗪基)苯并呋咱(7a)的合成 50 mL反应瓶,加入6(130 mg, 0.64 mmol)、20 mL无水乙醇和三乙胺(0.1 mL, 0.72 mmol),搅拌至全溶,缓慢滴加5.0 mL 1-甲基哌嗪(130 mg, 1.28 mmol)的无水乙醇溶液,室温反应14 h,反应完全。抽滤,室温干燥,得淡黄色固体7a(113 mg),收率18.9%,mp: 203~205 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.10(1H, d,J=10.0 Hz, 7-H-Ar), 7.85(1H, d,J=10.0 Hz, 6-H-Ar), 3.49(4H, t,J=4.44 Hz, piperazine-2, 6-H), 2.58(4H, t,J=4.40 Hz, piperazine-3, 5-H), 2.40(3H, s,-CH3)。ESI-MS,m/z: 221.0[M+H+]。参照化合物7a的合成方法,合成化合物7b—7e,其外观、总收率、熔点、1H NMR和ESI-MS 数据如下:
4-硝基-5-己胺基苯并呋咱(7b):黄绿色粉末,收率28.2%,mp: 83~84 ℃。1H NMR(400 MHz, DMSO-d6)δ: 10.67(1H, s,-NH-), 8.21(1H, d,J=10.0 Hz, 7-H-Ar), 7.74(1H, d,J=10.0 Hz, 6-H-Ar), 3.70(2H, dd,J=6.84, 13.88 Hz,-CH2CH2CH2CH2CH2CH3), 1.63~1.70(2H, m,-CH2CH2CH2CH2CH2CH3), 1.30~1.39(6H, m,-CH2CH2CH2CH2CH2CH3), 0.86(3H, t,J=6.84 Hz,-CH3)。ESI-MS,m/z: 287.06[M+H+]。
4-硝基-5-(4-异丙基哌嗪基)苯并呋咱(7c):黄色粉末,收率20.7%,mp: 169~171 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.07(1H, d,J=10.0 Hz, 7-H-Ar), 7.87(1H, d,J=10.0 Hz, 6-H-Ar), 3.52(4H, t,J=4.36 Hz, piperazine-2, 6-H), 2.66(4H, t,J=4.38 Hz, piperazine-3, 5-H), 2.69~2.76(1H, m,-CH-), 1.0(6H, t,J=7.36 Hz,-(CH3)2)。ESI-MS,m/z: 292.1[M+H+]。
4-硝基-5-(4-异丁基哌嗪基)苯并呋咱(7d):橘黄色粉末,收率21.3%,mp: 186~188 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.09(1H, d,J=10.0 Hz, 7-H-Ar), 7.89(1H, d,J=10.0 Hz, 6-H-Ar), 3.54(4H, t,J=4.64 Hz, piperazine-2, 6-H), 2.50~2.54(4H, m, piperazine-3, 5-H), 2.09(2H, d,J=7.40 Hz,-CH2-), 1.75~1.82(1H, m,-CH-), 0.87(6H, d,J=6.52 Hz,-(CH3)2)。ESI-MS,m/z: 306.1[M+H+]。
4-硝基-5-高哌嗪基苯并呋咱(7e):棕色粉末,收率27.4%,mp: 95~97 ℃。1H NMR(400 MHz, DMSO-d6)δ: 8.01(1H, d, J=10.0 Hz, 7-H-Ar), 7.79(1H, d,J=10.0 Hz, 6-H-Ar), 4.03(2H, s, homopiperazine-2-H), 3.89(2H, s, homopiperazine-7-H), 2.70~2.73(2H, m, homopiperazine-3-H), 2.49~2.52(2H, m, homopiperazine-5-H), 2.28(3H, s,-CH3), 1.99~2.02(2H, m, homopiperazine-6-H)。ESI-MS,m/z: 264.05[M+H+]。
1.3 体外抗肿瘤活性评价
将对数生长期的NCI H460、MCF-7及 HCT-116细胞,用5%培养基分别配置成浓度为4×104、3.5×104和 2×104个/mL的细胞悬液,并接种到96孔细胞培养板,每孔200 μL,培养24 h后去除原培养基,加入200 μL含5 μmol/L各目标物的培养基,每个浓度设4个平行孔;同时设阳性对照药5-Fu组(测定对NCI H460的抑制率的浓度为500 μg/mL,对MCF-7、HCT-116的抑制率的浓度为200 μg/mL)和阴性对照组(0.1% DMSO)各6个平行孔。继续培养48 h,每孔加入50 μL 50%TCA固定1 h,用去离子水冲洗5遍,甩干;干燥后加入100 μL SRB(4 mg/mL),室温染色15 min,弃去染色液,用1%乙酸冲洗5次;干燥后每孔加入150 μL Tris-Bace溶液(10 mmol/L),水平摇床上振荡20 min,用酶标仪测定540 mm下的吸光度A540。抑制率=[(A540对照孔-A540给药孔)/A540对照孔]×100%。并测定对肿瘤细胞MCF-7及HCT-116有较好抑制作用的目标物4a—4eIC50值。
2 结果与讨论
2.1 目标物的合成
首先以2-硝基-4-氯苯胺为原料环合得到氧化苯并呋咱环,然后在无水乙醇中,室温条件下,4-硝基-5-氯-苯并氧化呋咱(中间体3)与仲胺、4-取代哌嗪和取代(高)哌嗪通过亲核取代反应,得到目标化合物4a—4e。随着反应的进行,逐渐生成较多固体,TLC检测发现,生成的杂质较多,反应完全后,抽滤,取少量固体,用甲醇溶解,TLC检测,发现滤饼与母液的杂质相同,因此将溶剂蒸除,与固体混合,经氯仿萃取、干燥和硅胶柱层析,得到目标物。
纯化后的4a—4e通过1H NMR确证结构,核磁图谱显示产品含有一定比例的异构体。取代基为脂肪胺的化合物4b及取代基为甲基高哌嗪的化合物4e的异构体含量为35%~39%,取代基为取代哌嗪的目标化合物4a、4c及4d的异构体含量少于10%。TLC仅显示一个点,推测两个异构体的极性相同或者相近。有文献[11-12]报道,当温度达到5-取代-4-硝基-苯并氧化呋咱的熔点时,该化合物会发生重排,生成4-取代-7-硝基-苯并氧化呋咱,当取代基为给电子基团时,室温条件下也会发生重排。因此,以上结果与文献报道一致。
目标化合物7a—7e则由相应的苯并氧化呋咱,以三苯基膦作还原剂反应得到。
2.2 体外抗肿瘤活性评价
SRB法初步评价目标化合物4a—4e及7a—7d的体外抗肿瘤活性,结果见表1。
表1 4a—4e及7a—7d对NCI H460、MCF-7和HCT-116细胞的体外增殖抑制率
药理活性评价结果表明,浓度为5.0 μmol/L时,化合物4a—4e对NCI H460、MCF-7及HCT-116具有较强的抑制作用,多数化合物抑制率大于85.0%。7a—7d对三株肿瘤细胞的体外抑制活性较差,多低于50.0%。测定了对MCF-7及HCT-116两肿瘤细胞有较好抑制作用目标物的 IC50值,4a—4e对MCF-7的IC50值小于1.0 μmol/L,对HCT-116的IC50值小于5.0 μmol/L,优于先导化合物XI-006(IC50, MCF-7=5.8 μmol/L、IC50, HCT-116=15.3 μmol/L)。
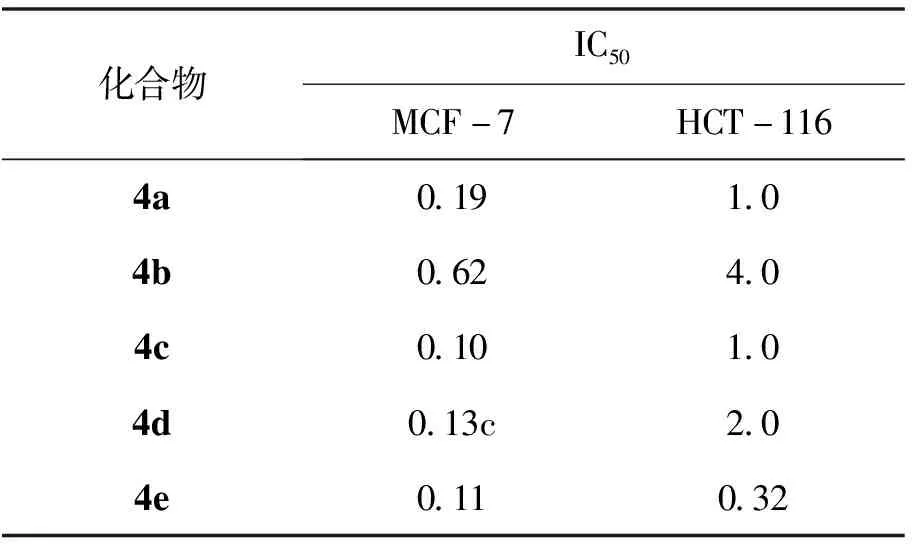
表2 4a—4e对MCF-7及HCT-116肿瘤细胞的IC50
3 结 论
本研究共合成了10个 4,5-二取代苯并(氧化)呋咱类新衍生物。结果表明,浓度为 5.0 μmol/L 时,化合物4a—4e对人非小细胞肺癌细胞NCI H460、乳腺癌细胞MCF-7和结肠癌细胞HCT-116具有较强的抑制作用,多数化合物的抑制率大于85.0%;化合物7a—7d对三株肿瘤细胞的体外抑制活性较差,多低于50.0%,表明苯并氧化呋咱类似物的抗肿瘤作用较苯并呋咱类似物强。4a—4e对MCF-7的IC50值小于1.0 μmol/L,对HCT-116的IC50值小于5.0 μmol/L,优于先导化合物XI-006。因此,4,5-二取代苯并氧化呋咱类衍生物具有进一步研究的价值。
猜你喜欢
中国药理学与毒理学杂志(2022年7期)2022-10-17
云南化工(2022年2期)2022-03-18
动物营养学报(2022年1期)2022-02-20
家庭医药(2021年8期)2021-07-28
父母必读(2021年3期)2021-02-04
分析化学(2017年12期)2017-12-25
成长·读写月刊(2017年11期)2017-11-25
今日农药(2014年7期)2014-09-15
