
黑龙江省种植大豆品种遗传多样性分析及与性状关联SSR标记筛选
2021-05-26 08:40李志江牛江帅鹿保鑫张东杰阮长青
中国粮油学报 2021年4期
李志江 牛江帅 李 忍 姜 鹏 鹿保鑫 张东杰 阮长青,2
(黑龙江八一农垦大学食品学院1,大庆 163319)(北大荒现代农业产业技术省级培育协同创新中心2,大庆 163319)(黑龙江省杂粮加工及质量安全工程技术研究中心3,大庆 163319)(黑龙江八一农垦大学生命科学技术学院4,大庆 163319)
黑龙江省是我国重要的大豆产区,脂肪和蛋白质等专用型大豆品种较多,但存在品种繁多乱杂、越区种植和混合种植等现象,严重影响了黑龙江省大豆的产量和质量,导致品牌保护难、种植和食用安全性低等问题[1]。传统的大豆品种鉴定,主要采用田间种植鉴定方法,但该方法时间长、效率低、工作量大、结果准确性低,所需鉴定目标性状大都为数量性状,受环境和品种同质化影响较大,给大豆品种的准确鉴定带来一定困难[2,3]。在分子鉴定水平上,DNA指纹图谱技术则具有高效准确、不受环境条件影响和实验操作简单等优点,已广泛应用于植物品种真实性鉴定及纯度检测研究[2]。目前,广泛应用的DNA分子标记主要包括限制性片段长度多态性(RFLP)、随机扩增多态性DNA(RAPD)、扩增酶切片段长度多态性(AFLP)、简单序列重复长度多态性(SSR)和单核苷酸多态性(SNP)等。其中,SSR分子标记具有数量丰富、多态性高、遗传共显性、谱带扩增稳定、精度高、检测时间短和技术成熟等特点,已广泛应用于植物品种鉴定及纯度检测的研究[4]。
目前,国内已有学者采用SSR标记技术鉴定大豆品种并构建指纹图谱,如徐海风等[5]利用6对SSR引物对淮河以南地区26份菜用大豆品种(系)构建指纹图谱,将26份菜用大豆品种(系)逐一区分。高运来等[6]利用9对SSR引物,完全区分黑龙江省83份参试大豆品种并构建了一套大豆品种的分子ID。何琳等[7]利用6对SSR引物鉴定了长江流域区域的42份参试大豆品种,并获得唯一的分子ID。这些数据均为满足大豆品种真实性鉴定的需求奠定了基础,但采用SSR标记并结合大豆群体关联分析鲜有报道。本研究的目的是采用分子标记技术,对黑龙江省75个大豆品种进行遗传多样性分析,并筛选与性状关联的SSR标记,为大豆种质资源真实性鉴定提供基础数据,为保障黑龙江省优质大豆种植和食用安全提供参考。
1 材料与方法
1.1 供试大豆
选用黑龙江省10个农场和地区的75份品种为参试材料,具体信息见表1。
1.2 大豆DNA提取
取发苗至三叶一心期大豆叶片于2 mL离心管内,-20 ℃过夜,加1 mL DNA提取液研磨成匀浆后,60 ℃水浴1 h,于12 000 r/min条件下离心5 min,取上清液备用。在上清液中加入等体积的氯仿,剧烈振荡,再次离心取上清液。重复1次后,加入等体积的异丙醇,-20 ℃放置1 h,于12 000 r/min条件下离心5 min,弃上清液后加入500 μL 70%乙醇洗2次,风干。加入100 μL TE缓冲液溶解,用于毛细管电泳检测。
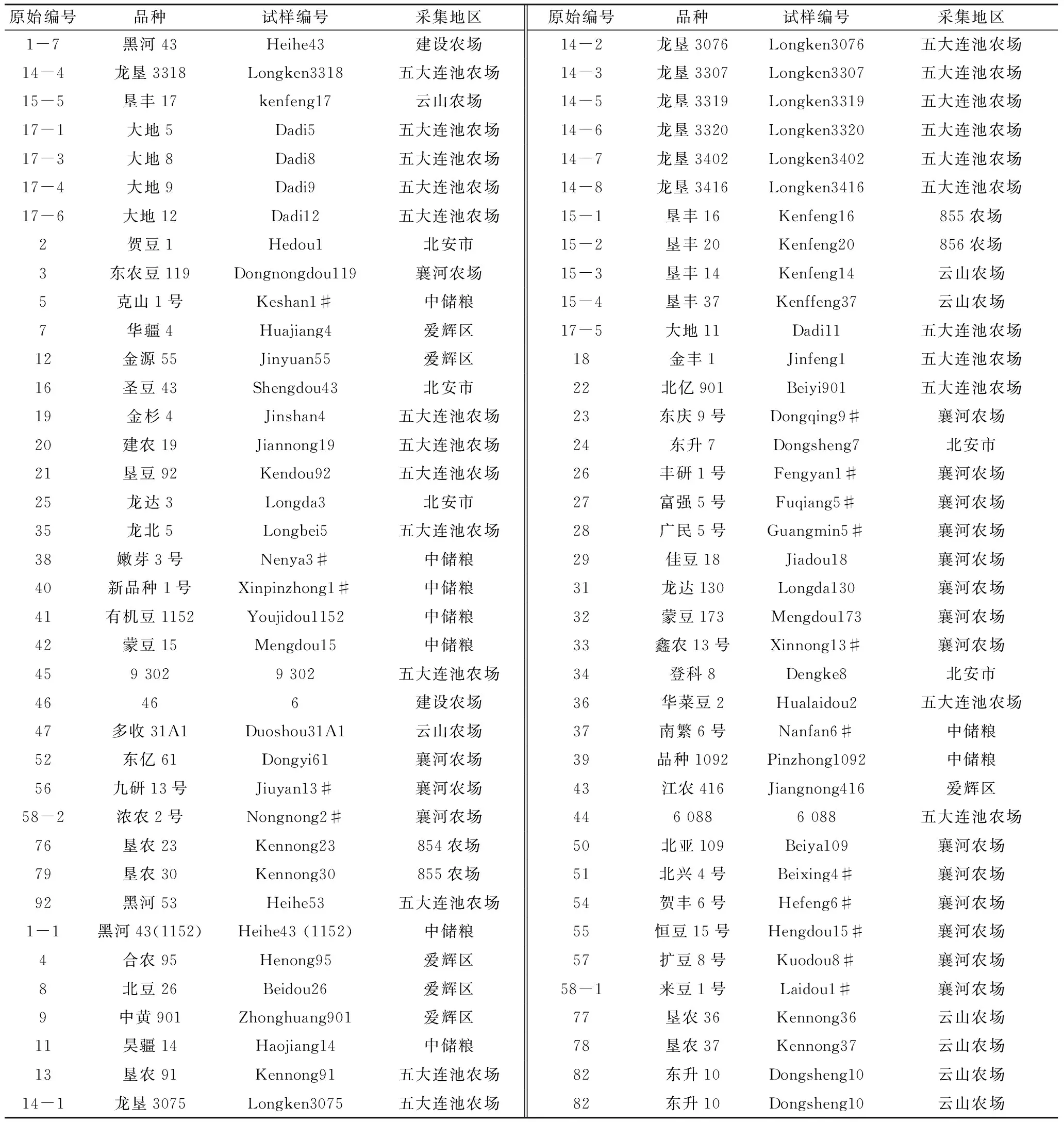
表1 黑龙江省10个农场和地区的75份品种试样信息

表2 SSR引物信息表
1.3 PCR扩增及毛细管电泳检测
从已发表文献中选出多态指数高的SSR标记引物,分布在15条染色体上的18对引物(如表2)。18对SSR标记引物序列来源于Soybase(https://soybase.org/dlpages/),由检测公司完成PCR扩增及毛细管电泳检测。正式实验前,随机选取6个样本对引物进行预实验,通过琼脂糖电泳确定是否有目的扩增产物,从而确定引物是否可用。
1.4 数据处理
1.4.1 SSR毛细管电泳有效特异峰的判定方法
扩增中容易出现影子峰干扰的问题,而影子峰是由Taq酶和SSR的特性决定的,基本上难以克服。杂合基因型的二倍体在理想情况下应该是扩增出2条等高的峰,但由于受扩增条件、加样的随机误差、DNA模板质量及DNA浓度等因素的影响,致使扩增出的两峰不等高。对于杂合型的二倍体有效特异峰的判断,采用小峰面积如果不及大峰面积的60%则判定为杂带的标准,对于通过优化扩增条件的方法难以改善,以及无法判读的位点进行舍弃。
1.4.2 标记基因型的数据分析
通过PowerMarker V3.0得到每个SSR分子标记的多样性信息[8],如多态性百分比、最小等位基因频率/位点、等位基因数/位点、有效等位基因数/位点、等位基因型数/位点、基因多样性/位点、多态性信息含量/位点和多样性指数/位点等,基于位点多样性信息的平均水平来评估群体遗传多样性。
1.4.3 群体关联数据分析
利用Structure V2.3.4软件进行群体结构分析[9]。参数设置为:K值选取1~10,重复次数为5;将MCMC(Markov chain monte carlo)开始时的不作数迭代设为100 000次,再将不作数迭代后的MCMC设为1 000 000次,其余参数采用软件默认的设置。根据lnP(D)计算ΔK,依据ΔK值选择一个合适的K值,并得到该K值对应的Q矩阵。将Clump软件分析的结果传递给Distruct软件生成Q值条形堆积图。然后利用Tassel V2.1软件[10]将基因型数据生成亲缘关系矩阵(K矩阵),结合等位变异数据、基因型数据、各个环境的表型值、Q矩阵,利用混合线性模型MLM(Mixed linear model)进行性状和标记之间的关联分析,并计算标记位点在P<0.05和P<0.01时对表型变异的贡献率(R2)。在已获得关联位点的基础上,再进行优异等位变异的发掘,通过对SSR位点等位变异表型效应计算,最终获得与表型性状显著关联的位点。最后通过PowerMarker V3.0计算基于等位基因频率的遗传距离,采用邻位相连(Neighbor-Joining, NJ)法进行聚类分析,并输出聚类树[11]。
2 结果与分析
由于SSR扩增中容易出现影子峰干扰以及非特异峰的问题,需要对有效特异峰进行判定。以Satt233标记的结果为例,在图1a中,箭头所指的峰为有效特异峰;图1b中,箭头所指的2个峰面积接近,且小峰面积超过大峰面积的60%,视为有效特异峰。

表3 42个大豆品种二进制身份证


图1 SSR扩增片段有效特异峰型的判定
2.2 大豆品种指纹编码的构建
根据毛细管电泳检测结果,对每个SSR分子标记有效谱带的“有”或“无”采用“1”或“0”进行系统记录,构建大豆品种的二进制指纹编码。构建了10个单一SSR标记区分42个大豆品种的身份证(表3),其中Satt100和Sat_128的鉴别能力最强,分别能够鉴定出7个品种。个别品种能够被多个标记鉴定出来,如北亿901和6 088。另33个大豆品种需多个SSR标记共同区分。
2.3 群体遗传多样性分析
采用18个SSR分子标记对75份大豆品种进行多样性分析,各位点等位变异的数目从3(Satt387)~12个(Satt100),平均每个位点等位变异数为7.333个。各位点多态信息量(PIC)从0.146(Sat_084)~0.675(Satt442),平均多态信息含量为0.405(表4)。其中,PIC可以用来衡量基因变异程度的高低,一般认为,当PIC<0.25 时,为低度多态性信息引物,当0.25

表4 大豆遗传多样性分析统计表
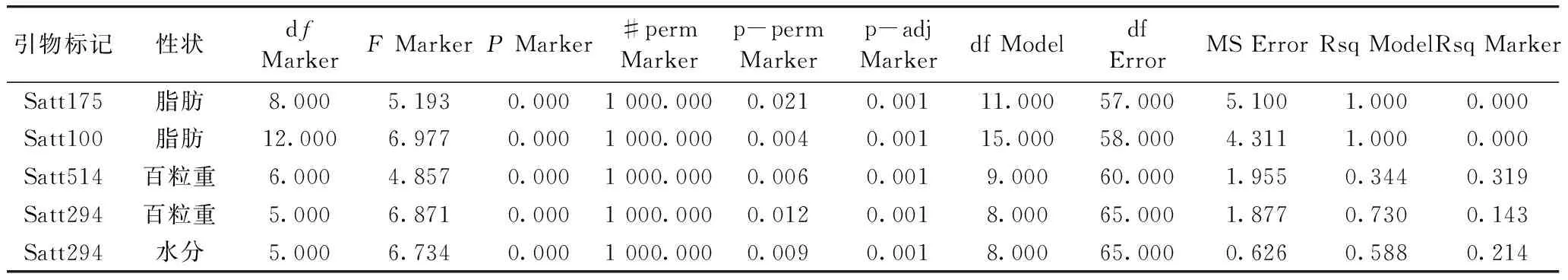
表5 相关性状与标记关联情况
2.4 群体结构分析
通过Structure软件对75份大豆品种的种群结构分析结果显示,最显著的ΔK变化出现在K从2~3时,峰值出现在K为3时(图2a)。根据确定的K值和Clump合并的Q矩阵,经Cluster软件分类后生成Q值条形堆积图。不同的序号区域表示不同的“血统”,或意味着该大豆来源于不同的祖先群体。结果显示,本研究的大豆群体大体上可以分为3个亚群,而种群内部,品种间存在着基因频率的差异(图2b)。
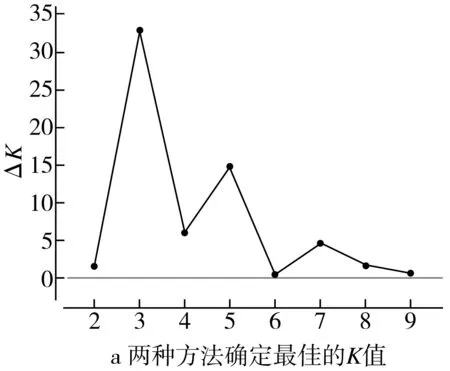
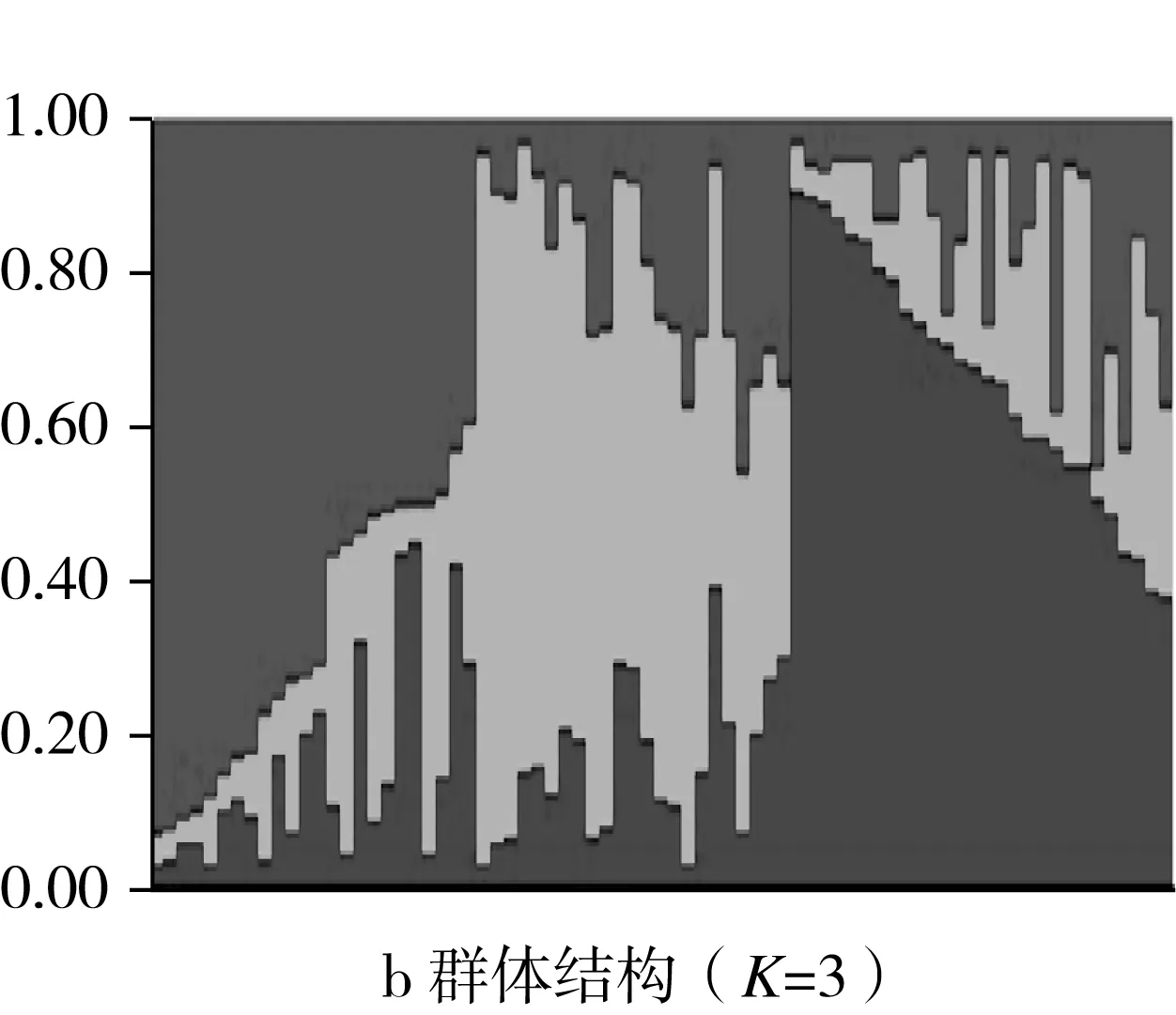
图2 大豆群体遗传结构分析
2.5 性状的位点关联分析
在群体结构分析结果中,选取K=3的10个重复中P值较大的Q矩阵进行关联分析。利用Tassel V2.1软件,采用混合线性模型(MLM)对18个SSR标记与4个大豆相关性状进行关联分析。结果显示,共鉴定出与性状显著关联的5个SSR标记,涉及了4个SSR标记(表5)。其中,2个SSR标记(Satt175和Satt100)与脂肪存在显著关联,标记主要分布于6和7号染色体上;2个SSR标记(Satt514和Satt294)与百粒重存在显著关联,标记主要分布于4和17号染色体上,其中Satt294与百粒重和水分均存在显著关联。不同大豆品种在百粒重、蛋白质、水分和脂肪性状上的平均值为(18.94±1.74) g、34.67%±0.93%、10.24%±0.78%和18.2%±0.55%,变异系数分别为0.159、0.046、0.132和0.053。
3 讨论
目前,虽然利用SNP进行基因分型的研究较多,且在基因组上SNP比SSR数量多,但是与SNP相比,SSR的突变频率会高于SNP且蕴含的信息也更丰富。所以目前SSR广泛的用于基础和应用研究领域中[13]。关于大豆遗传多样性的研究很多[6,7,14-17]。在本研究中,选取的18对多态性较高的SSR引物中10个单一SSR标记可区分42个品种,明显低于高运来等[6]筛选出的9对引物和徐冬雪等[14]筛选出的3对引物所鉴别出来的品种数量,说明本研究中所涉及75个品种的遗传多样性相对较低。
为进一步揭示该研究群体大豆的群体分化与亲缘关系,基于Nei’s遗传距离利用邻位连接法(Neighbor-Joining)构建了图3的系统发育树。对比基于Structure软件和 NJ聚类2种方法进行群体结构分析的结果,2种方法均显示75份大豆基因型基本上可以分为3个群体,但是2种方法分出的3个亚群组成上有所差异。造成这种差异的主要原因是两者分类的依据不同,相比之下,基于数学模型的群体遗传结构分类比基于材料间的遗传距离的聚类分析更加精确[18]。而群体结构划分不合理时,极易造成标记与性状之间的伪关联[19]。本研究中利用Structure软件将材料分为了3个群体,这样可以更加精确地进行关联分析。
关联分析是目前国际植物基因组学研究中发掘作物优异基因的重要方法之一,是利用随机且均匀分布于基因组的标记信息来估计群体内部个体间的遗传关系,并作统计假设检验进行基于群体的关联分析[19]。该分析不需要构建群体和遗传图谱,省去了大量的工作,同时也可以通过该方法发现优异基因,为遗传育种提供依据[18]。本实验用Tassel V2.1软件采用MLM混合模型对18个SSR标记与4个大豆农艺性状进行了关联分析。MLM 混合模型较好地控制了一般线性模型(General linear model,GLM )的错误,降低了假阳性的概率[19]。本研究中发现了4个与性状相关联的标记,Satt175、Satt100、Satt514和Satt294。王海滨[20]利用已定位的6对与大豆蛋白质和脂肪含量相关的SSR标记对180份大豆材料进行高油检测时,发现Satt100检测符合度可以达到87.80%,与本研究结果一致。
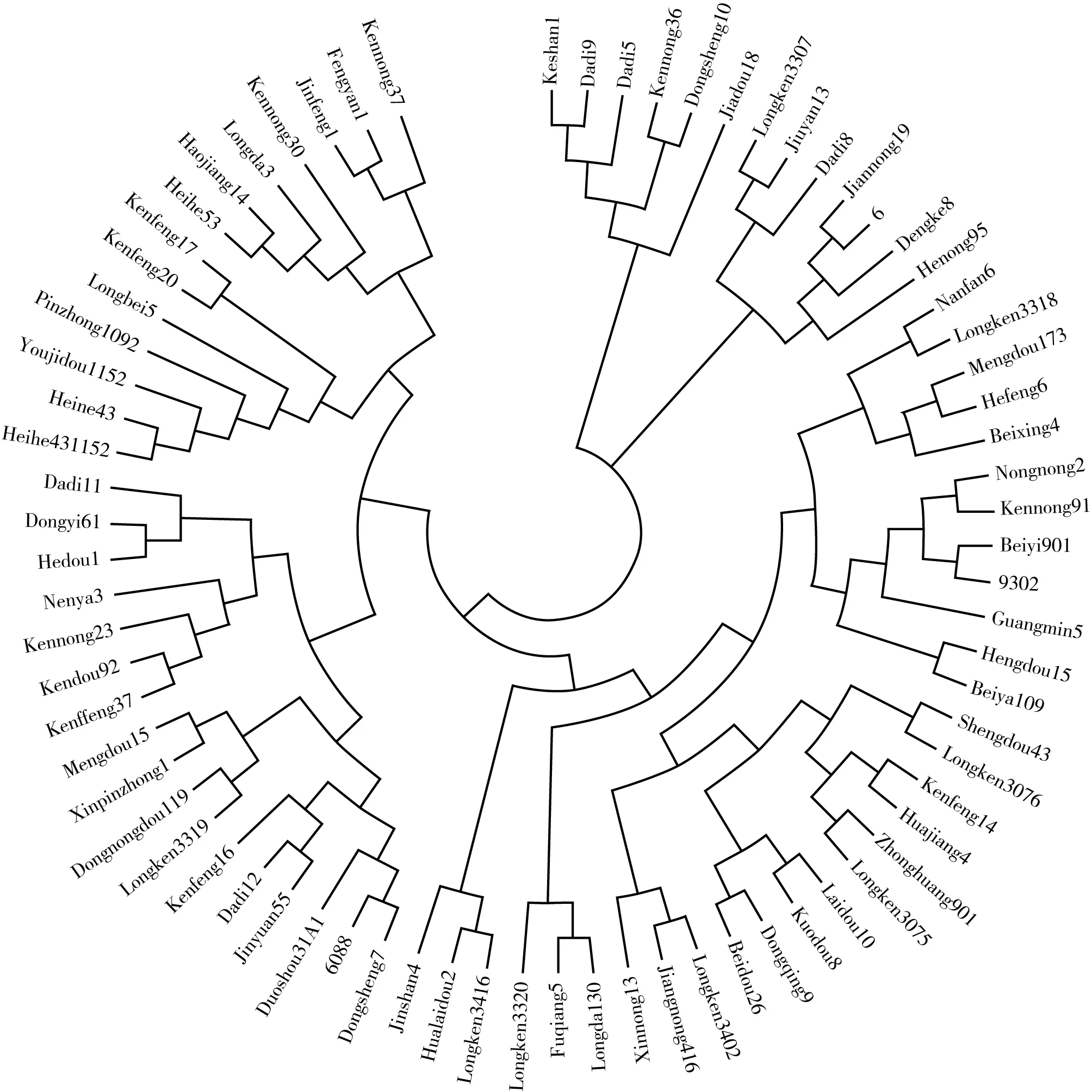
图3 基于Nei’s遗传距离绘制的NJ树
4 结论
通过SSR分子标记技术对黑龙江省75份大豆品种进行遗传多样性、群体遗传结构以及性状与分子标记关联分析,结果显示,各位点等位变异的数目变化从3(Satt387)~12个(Satt100),平均每个位点等位变异数为7.333个,各位点多态信息量变化从0.146(Sat_084)~0.675(Satt442),平均多态信息含量为0.405。并初步构建了10个单一SSR标记鉴定42份大豆品种(系)的DNA指纹图谱。群体结构分析将该研究群体分为3个亚群。通过关联分析共鉴定到与脂肪、百粒重和水分等性状相关的4个SSR标记,分别为Satt175、Satt100、Satt514和Satt294。最终基于Nei's(1973)遗传距离,构建了75份材料的系统进化树,明确品种间亲缘关系远近。
猜你喜欢
今日农业(2022年16期)2022-11-09
中国化肥信息(2022年5期)2022-08-30
世界科学技术-中医药现代化(2022年3期)2022-08-22
今日农业(2021年20期)2021-11-26
新世纪智能(数学备考)(2021年9期)2021-11-24
今日农业(2021年14期)2021-10-14
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
读者(2017年5期)2017-02-15
西南农业学报(2016年6期)2016-04-16
