
Differentially expressed genes analysis and target genes prediction of miR-22 in breast cancer*
2021-05-22 11:26:12TaoFanChaoqiWangCofirstauthorKunZhangHongYangJuanZhangWanyanWuYingjieSong
Tao Fan,Chaoqi Wang (Co-first author),Kun Zhang,Hong Yang,Juan Zhang,Wanyan Wu,Yingjie Song (✉)
1 Department of Oncology,The People's Hospital of China Three Gorges University,The First People's Hospital of Yichang,Yichang 443000,China
2 Department of Urinary Surgery,Affiliated Hospital of Inner Mongolia University for the Nationalities,Tongliao 028007,China
3 Department of Orthopedics,The People's Hospital of China Three Gorges University,The First People's Hospital of Yichang,Yichang 443000,China
4 Department of General Surgery,The People's Hospital of China Three Gorges University,The First People's Hospital of Yichang,Yichang 443000,China
Abstract Objective miR-22 is highly active in breast cancer,especially in the luminal B and HER2 subtypes.However,the detailed potential of the use of target genes for miR-22 in breast cancer are still unclear.In this study,we aimed to discover potential genes and the miRNA-DEGs network of miR-22 in breast cancer using bioinformatics approaches.Methods Analysis of microarray data GSE17508 (including 3 miR-22 knockout samples and 3 controls)obtained from the Gene Expression Omnibus (GEO) database was performed.Differentially expressed genes (DEGs) between the miR-22 knockout samples and the three control samples were detected using GEO2R.The gene ontology (GO) functional enrichment analysis and protein-protein interaction (PPI)network of DEGs were performed using the online tool Metascape and STRING database,separately.The miR-22 and DEG networks were obtained from the miRNet database.Cytoscape software was used to construct and analyze a merged miRNA-DEG network.The online tools database,mirDIP 4.1,was used to predict miR-22 target genes.Results Certain DEGs and miRNAs may be potential targets for predicting and treating miR-22 expressed breast cancer.Conclusion We constructed a prognostic model of rectal adenocarcinomas based on four immunerelated lncRNAs by analyzing the data based on TCGA database,with high prediction accuracy.We also identified two biomarkers with poor prognosis (PXN-AS1 and AL158152.2) and one biomarker with good prognosis (LINC01871).
Key words:bioinformatics;breast cancer;MCF7 cells;MiR-22
MicroRNAs (miRNAs) are a class of noncoding single-stranded RNA molecules with a length of about 22 nucleotides encoded by endogenous genes,which coordinate multiple gene expression programs through gene regulation[1-2].There are over 1,700 identified miRNAs in the human genome that are associated with a wide variety of human cancers,such as breast,lung,and colon cancer[2-3].Research suggests that miRNAs may be related to the pathogenesis of cancer,tumor growth,and metastasis,and play the role as oncogenes or tumor suppressor genes[4].Therefore,the identification of miRNA targets is considered to be key in improving our understanding of the regulatory effects of miRNAs.miR-22 has been identified as a regulator of lipid and folic acid metabolism in breast cancer cells through the systematic integration of the molecular spectrum[5].Songet al.found that miR-22 regulated breast cancer stemness and metastasis via TET (ten eleven translocation) family dependent chromatin remodeling[6].Experiments and clinical studies show that miR-22 promoted epithelial mesenchymal transition and tumor invasion and metastasis[7].Hence,exploring miR-22 target genes is important for targeted therapy of breast cancer.
In this study,the differentially expressed genes (DEGs)from microarray data in the GEO database were identified between knockout or duplex miR-22 samples in human breast cancer MCF-7 cell lines.Online tools including mirDIP 4.1 software were used to predict miR-22 target genes.We aimed to explore certain differential genes,and miRNAs may be potential targets for predicting and treating miR-22 expressed breast cancer.
Materials and methods
Microarray data filtering eligible data set
We searched and downloaded microarray data GSE17508 (including three miR-22 knockout samples and three controls) from the GEO database (www.ncbi.nlm.nih.gov/geo/).The datasets used human breast cancer MCF-7 cell lines.Microarray data were used to analyze the eligible data set and identify the gene expression patterns.
DEGs’ screening
DEGs were independently screened using the GEO2R online tool in the GEO database.In our study,DEGs between miR-22 knockout samples and controls were screened and selected by the cut-off point of adj.Pvalue< 0.05 and |log FC|> 0.5.
Functional enrichment and protein-protein interaction (PPI) analysis
The functions of DEGs were further analyzed using the online tool Metascape (http://metascape.org/gp/index.html#/main/step1).The terms withP-value <0.01,minimum count of 3,and enrichment factor > 1.5 were collected and grouped into clusters based on their membership similarities.Furthermore,we obtained the PPI of differential genes using the Search Tool in the Retrieval of Interacting Genes database (STRING,http://string-db.org/),and one big pairing picture was generated.In this study,DEGs with a confidence score of > 0.4 were selected to construct the PPI network.
Established miRNA-DEG network
The DEGs and has-miR-22 were uploaded to the miRNet 2.0 database (www.mirnet.ca/faces/home.xhtml) to acquire a list of miRNA-DEG pairs,and one big pairing picture was generated.The picture was merged with the PPI picture,and a complete and huge miRNAs-DEG network was generated using the Cytoscape 3.6.1.software[8].Then,the Molecular Complex Detection(MCODE) plugin was used to module clustering analysis to detect the potential functional modules in the network.In the MCODE process,the cut-off value of degree was set to 2,and the cut-off value of node score was set to 0.2.
Predicted target genes
To obtain potential target genes,hsa-miR-22 and the DEGs were uploaded to the online tools database mirDIP 4.1 (http://ophid.utoronto.ca/mirDIP/index_confirm.jsp).Researchers can predict target genes of miRNA with the help of this online tools database.We only selected the gene symbol whose confidence class is considered very high.
Results
DEG screening between miR-22 knockout samples and controls
An assessment of data normalization and crosscomparability (Table 1),and then DEGs analysis was carried out.
We applied an online tool (http://www.heatmapper.ca/expression/) analysis to discern the differential expression of genes.A total of 40 DEGs were identified in the profiles,including 38 upregulated and 2 downregulated DEGs in breast cancer MCF7 cells (Fig.1).
Gene ontology and functional enrichment analysis
Pathway and process enrichment analysis was performed using GO Biological Processes,GO Cellular Components,GO Molecular Functions,KEGG Functional Sets,and KEGG Pathway Ontology sources.
Heatmap-selected GO showed a defense response to virus (GO:0051607) and interferon signaling(R-HSA-913531) being the top two pathways (Fig.2a).The heatmap-selected GO parent showed a multiorganism process (GO:0051704),immune system process(GO:0002376),and signaling (GO:0023052) as the top three pathways (Fig.2b).
Construction of miRNA-DEG network analysis
Taking the selected 40 DEGs and has-miR-22 into account,we identified 393 PPI pairs using the STRING database as well as a large network of 284 genes and 751 miRNAs using miRNet.Two big pairing pictures mentioned above were merged using Cytoscape software to obtain a complete miRNA-DEG network in Cytoscape.The MCODE plugin in Cytoscape was used to perform the module clustering of the miRNA-DEG network mentioned above,and then the key functional modules of the network were evaluated.
Two modules were identified and showed certain differential genes (Fig.3 and 4),such as PARP14 (Poly-adenosine diphosphate-ribose polymerase 14),SAMHD1(Sterile alpha motif and histidine/aspartic acid domaincontaining protein 1),CCL5 (C-C chemokine ligand 5),and TNFSF10 (Tumor Necrosis Factor superfamily 10).
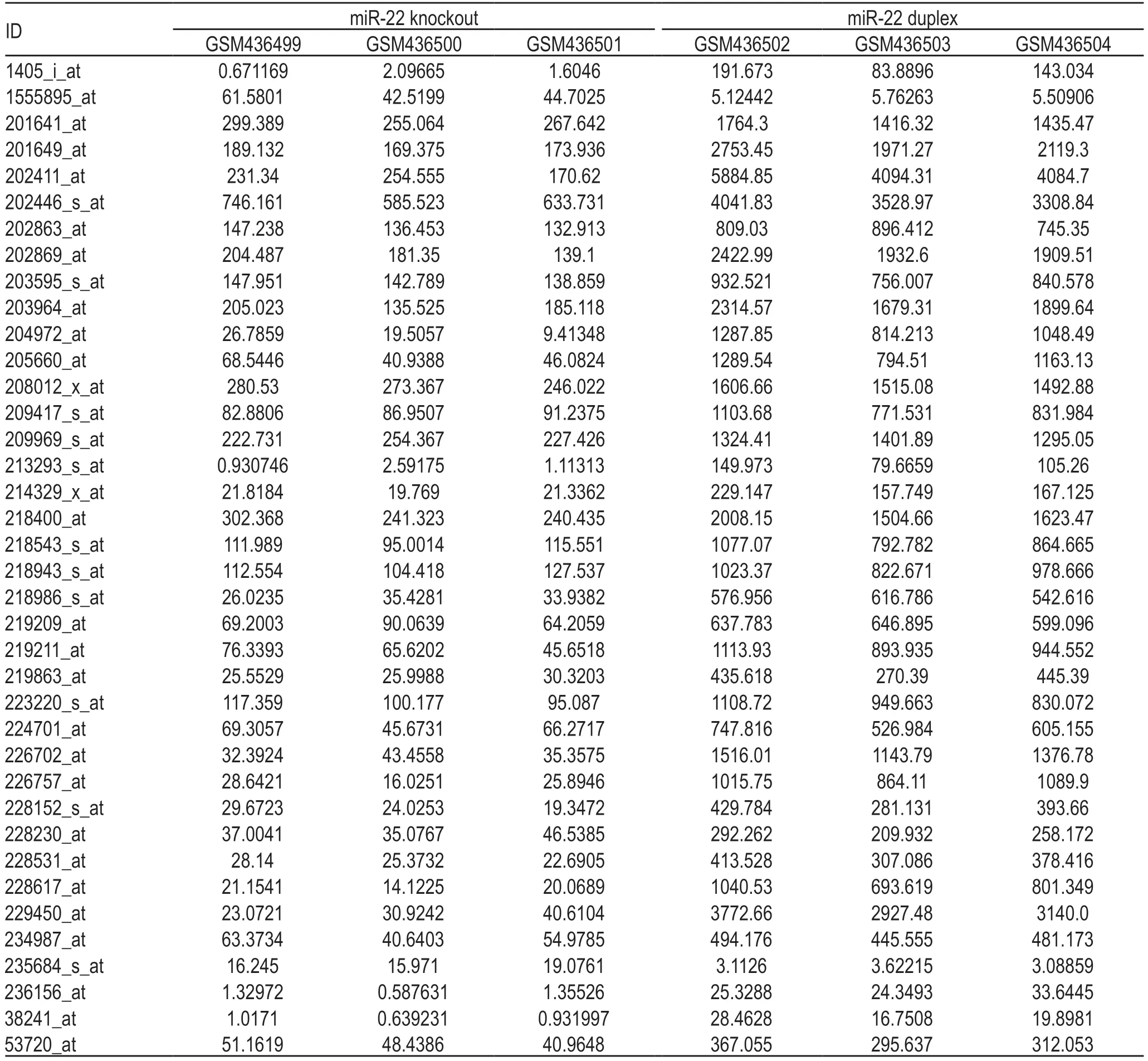
Table 1 Data normalization and cross-comparability
The four DEGs,PARP14,SAMHD1,CCL5,and TNFSF10,in these modules also occurred in the GO terms enriched above,which were associated with multiorganism immune processes and signaling pathways.PARP14 and SAMHD1 targeted miRNAs,such as miR-21-3p,miR-138-5p,miR-130a-3p,miR-155-5p,miR-452-5p,and miR-124-3p.We also observed a key hub,miR-146a-5p,that interacted with CCL5 and TNFSF10.
Predicted target genes
We identified that upregulated OAS1 and downregulated DEGs may be potential target genes of miR-22-3p.These were predicted using the online tools database mirDIP 4.1,with the confidence class selected as very high (Table 2).
Discussion
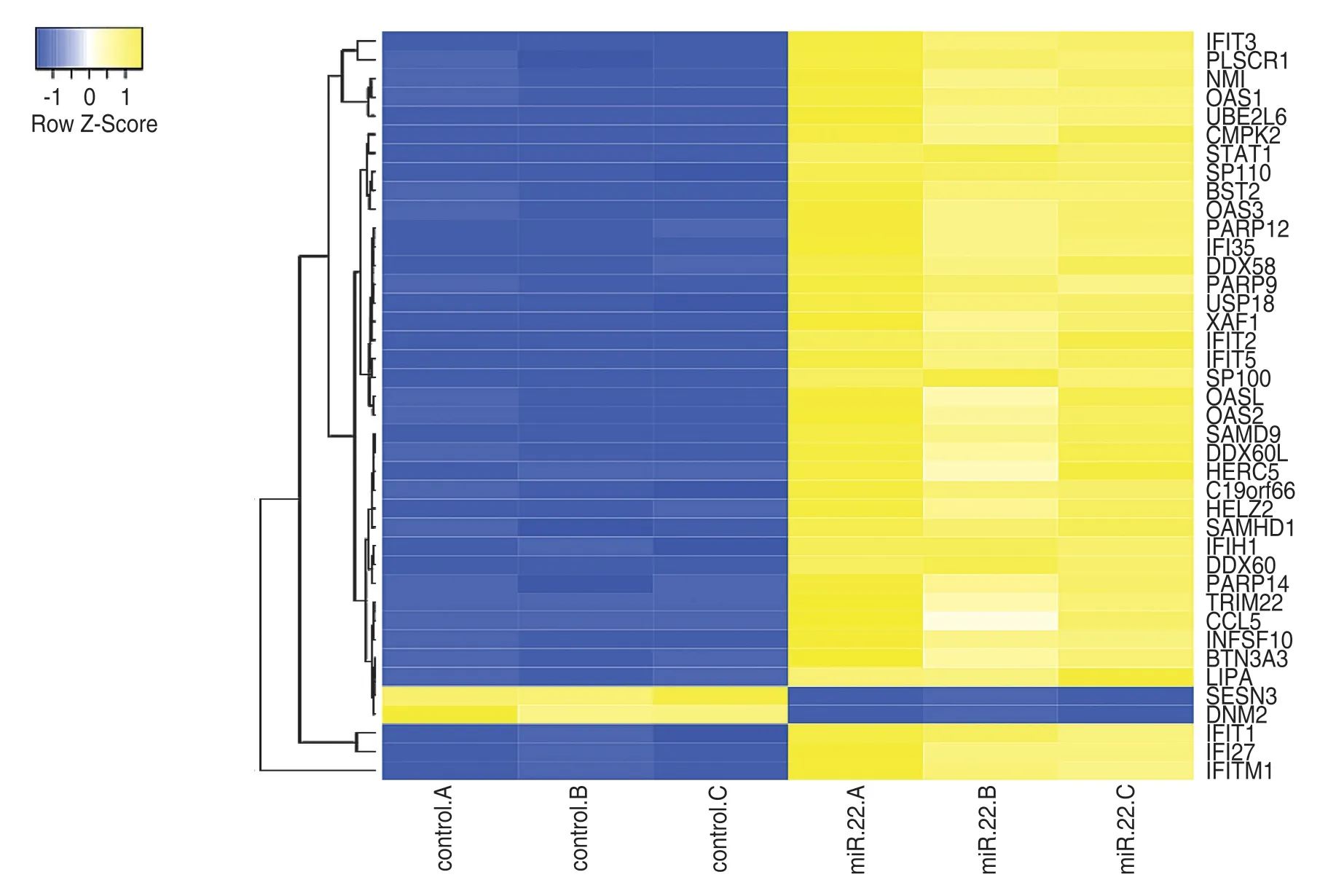
Fig.1 DEGs expression profiling in breast cancer MCF7 cells.
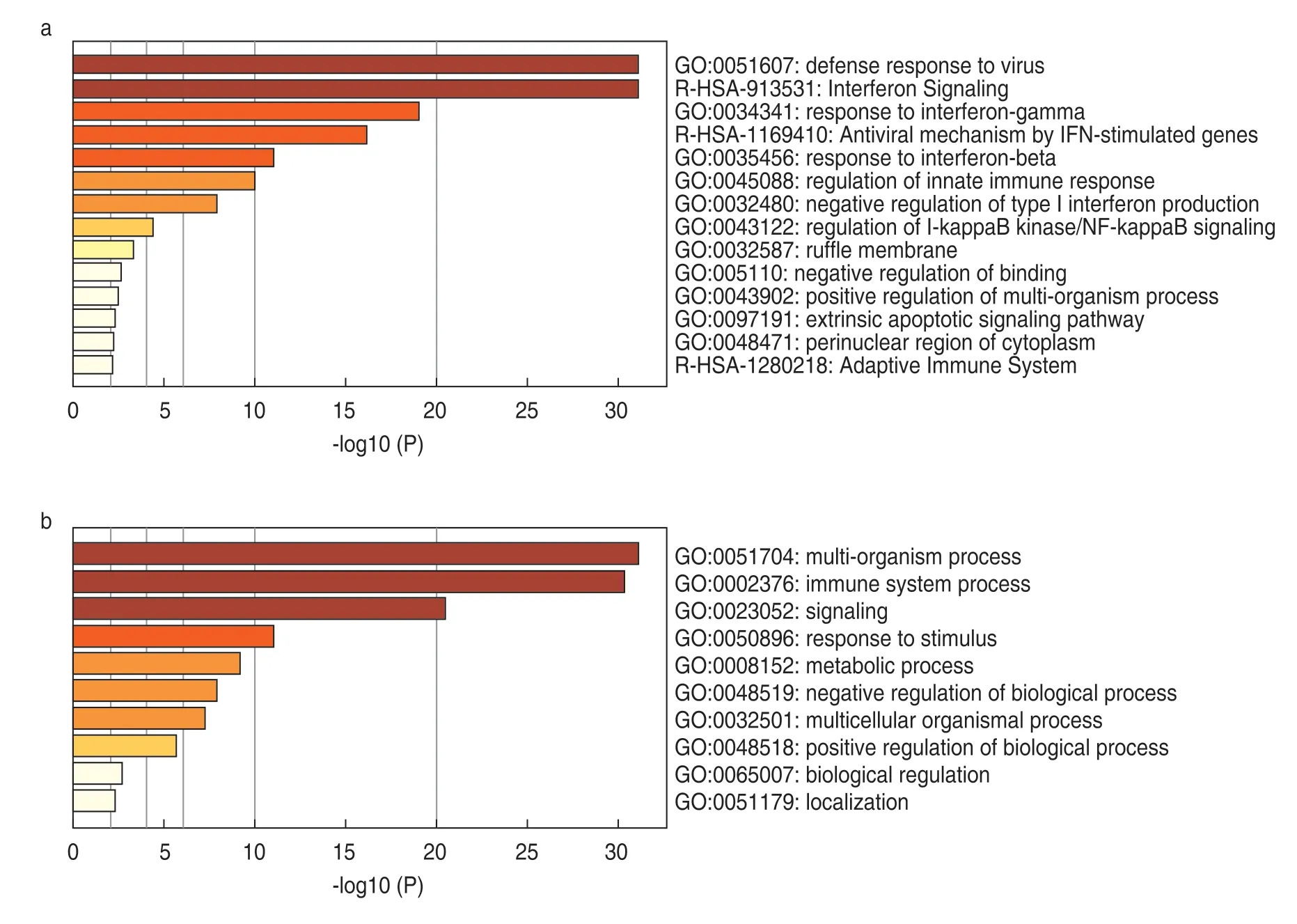
Fig.2 Distribution of gene ontology terms for the DEGs.(a) heatmap-selected GO;(b) heatmap-selected GO parent

Table 2 The predicted target genes of hsa-miR-22-3p

Fig.3 An outstanding hub of DEGs,PARP14 and SAMHD1,interacting with their miRNAs.
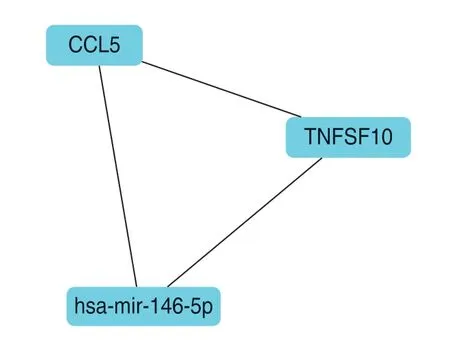
Fig.4 Another outstanding hub of DEGs,CCL5 and TNFSF10,interacting with miR-146a-5p.
Breast cancer is the most common malignant tumor in women worldwide,and metastasis is the main cause of death.Increasing evidence indicates that miR-22 is upregulated in breast tumors compared to its marginal non-tumor counterparts,which are associated with poor overall survival[9-10].However,the detailed potential target genes of miR-22 in breast cancer are still unclear.Thus,it is very important to study the molecular mechanism of breast cancer metastasis and progression to develop a therapeutic strategy for breast cancer patients.
As the miRNA-DEG network analysis showed,expression of miR-22 was closely related to the PARP14,SAMHD1,CCL5,and TNFSF10 genes in breast cancer MCF7 cells.PARP14 uses nicotinamide adenine dinucleotide (NAD+) as a metabolic substrate to modify the target protein by single ADP ribosylation,which is involved in cellular reactions and signaling pathways in the immune system.SAMHD1 is considered an intrinsic viral limiting factor that inhibits the process of viral infection,including retrovirus replication,packaging,and transmission.Several studies have demonstrated that PARP14 and SAMHD1 are associated with the development of multiple types of cancer,such as lung,colon,breast,myeloma,and pancreatic cancer among others[11-12].In the present study,through MCODE analysis,we found that PARP14 and SAMHD1 interact with some miRNAs,such as miR-21-3p,miR-138-5p,miR-130a-3p,miR-155-5p,miR-452-5p,and miR-124-3p.We found that miR-21-3p may be the core of the module because the survival analysis of miRNAs shows that the overexpression of miR-21 can significantly reduce the overall survival rate of breast cancer patients[13].
CCL5 is secreted by breast cancer cells and rarely expressed in epithelial cells of normal ducts or benign breast masses[14].Increasing studies showed that the CCL5/CCR5 axis is involved in tumor growth,migration and angiogenesis in different tumor types,including breast cancer.In addition,CCL5/CCR5 is closely related to the recruitment of tumor-associated immune-suppressive cells and promoting the construction of the tumor microenvironment[14].We found that CCL5 may also be the core of the module,through MCODE analysis,CCL5 interacts with TNFSF10 and miR-146a-5p.Moreover,in order to predict the target gene of miR-22 in DEGs,through the use of the online tools database mirDIP 4.1,we found that upregulated OAS1 and downregulated SESN3 may be potential target genes of miR-22-3p.
Conclusion
In summary,certain DEGs and miRNAs may be potential targets and biomarkers for predicting and treating miR-22 expressed breast cancer.However,further studies such as disease models and PCR experiments are necessary to verify these findings.
Conflicts of interest
The authors indicated no potential conflicts of interest.
 Oncology and Translational Medicine2021年2期
Oncology and Translational Medicine2021年2期
- Oncology and Translational Medicine的其它文章
- A case report of iodine-125 seed placement during operation for the treatment of advanced gallbladder carcinoma with septic shock*
- Analysis of the adverse reactions of atezolizumab:A real-world study based on FAERS database
- Analyses of the clinical characteristics of 49 cases of malignancy with multiple bone lesions as the first manifestation
- Application of endoscopic nasobiliary cutting in the treatment of hilar cholangiocarcinoma
- KIF15 expression characteristics:Relevance toneo-adjuvant chemotherapy efficacy in breast cancer*
- Study on the antitumor effects of autologous and allogeneic CIK cells in patients with breast cancer*