
一种2,3,5三取代噻吩的合成方法
2021-05-21 02:14范威
复旦学报(自然科学版) 2021年2期
范 威
(滁州城市职业学院 科研处,安徽 滁州 239000)
噻吩环存在于天然产物、具有生物活性的物质、合成中间体等有机化合物中[1],该类衍生物可用作PI3K抑制剂[2]、LRRK2抑制剂[3]、17β-HSD1抑制剂[4].如图1所示,舒洛芬(Suprofen)[5]、氯吡格雷(Clopidogrel)[6]、噻美尼定(Tiamenidine)[7]等活性分子均含有噻吩骨架.按照该骨架上取代基的数目,可将其分为单取代噻吩、双取代噻吩和多取代噻吩.舒洛芬属于单取代噻吩,具有抗炎活性.氯吡格雷属于双取代噻吩,具有抑制血小板凝集的作用.噻美尼定属于多取代噻吩,具有抗高血压的功能.除此之外,噻吩衍生物因其结构刚度和电子性能而被广泛用作导电有机材料[8].该骨架包括1个硫原子和4个碳原子,按照原子的来源可将其合成方法分为[4+1]法[9]、[3+2]法[10]、[2+2+1]法[11]等不同的形式.

图1 含有噻吩骨架的活性分子Fig.1 Active molecules with thiophene skeleton
Li等[12]用正丁基锂(n-BuLi)催化卤代噻吩和硼酸三异丙酯,制得了单取代噻吩.Connor等[13]从1,4-二噻吩-2,5-二醇和硝基烯烃出发,经两步反应合成了双取代噻吩.Nandi等[14]以4-二甲基氨基吡啶(DMAP)催化β-酮二硫酯和炔酸酯,经[3+2]环加成过程,实现了多取代噻吩的合成.
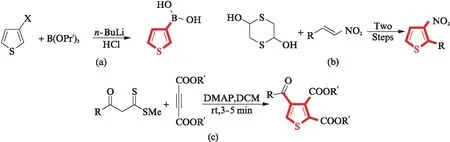
图2 噻吩骨架的修饰Fig.2 Modification of thiophene skeleton
多取代噻吩[15]包括2,3,4-三取代噻吩、2,3,5-三取代噻吩、2,3,4,5-四取代噻吩.其中,2,3,5-三取代噻吩可用作多种抑制剂,因此在噻吩衍生物中显得格外显眼.对其的合成探索也得到了持续研究.

图3 含有2,3,5-三取代噻吩的活性化合物Fig.3 Active compounds containing 2,3,5-trisubstituted thiophenes
Yue等[16]发展了2-炔基吡咯烷与硫的[4+1]环化反应,通过吡咯烷β-位的亲电进攻和随后的分子内噻吩环化过程,合成了2,3,5-三取代噻吩.该反应需要24 h,时间过长.Nakao等[17]在四氢呋喃(THF)溶液中用1,8-二氮杂二环十一碳-7-烯(DBU)催化丙二烯基酯和巯基乙酸甲酯,经历串联硫杂Michael和Dieckmann缩合过程,区域选择性合成了2,3,5-三取代噻吩.该反应的缺点是选用的原料巯基乙酸甲酯具有异味(图4(b)).Fang等[18]以二烷硫基烯炔为原料,氢氧化钠(NaOH)为催化剂,在80 ℃的二甲基亚砜(DMSO)溶液中反应,合成了19例2,3,5-三取代噻吩.该反应部分产物的分离产率有待提高(图4(c)).
本课题组以芳酰基取代的苊酮和硫氰基取代的芳酮为原料,叔丁醇钠(t-BuONa)为催化剂,在微波辐射条件下合成了2,3,5-三取代噻吩.与已有路线相比,该反应用时短、原料无味、产率高,无需金属催化剂,具有较高的实用性(图4(d)).
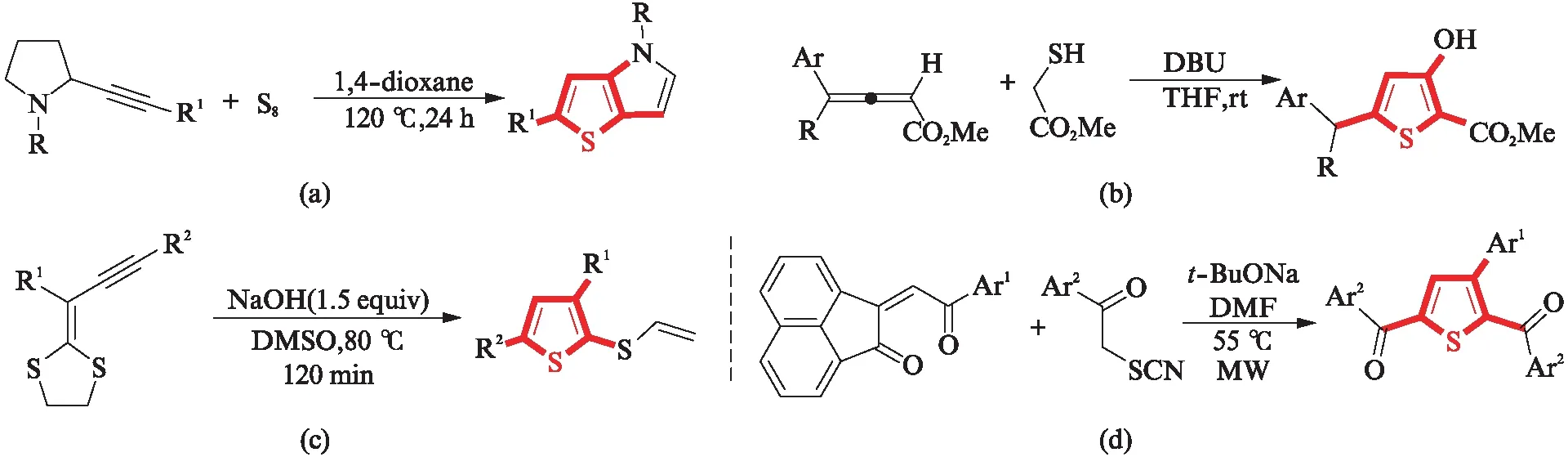
图4 噻吩骨架2,3,5-位的修饰Fig.4 Modification of 2, 3, 5-positions of thiophene skeleton
1 材料与方法
1.1 仪器与试剂
Monowave 200型微波合成仪;XT-5型显微熔点测定仪;FTIR-Tensor 27型红外光谱仪;DPX 400 MHz 型核磁共振仪;micrOTOF-QⅡ型质谱仪;Siemens P4型四圆衍射仪;所用试剂均为分析纯.
1.2 合成通法
将芳酰基取代的苊酮(0.5 mmol)、硫氰基取代的芳酮(1.0 mmol)、t-BuONa(0.5 mmol)投入到10 mL 规格的微波合成反应管中.加入2.0 mL的DMF,先混合均匀,然后盖上瓶盖,在微波合成仪中反应(55 ℃,20 min).将反应物取出冷却,倒入水中搅拌、减压抽滤、重结晶,最终得到2,3,5-三取代噻吩(产率66%~83%).
1.3 表征数据
3a白色固体; 熔点: 208~209 ℃;IR(KBr,v): 1 085,1 172,1 211,1 258,1 400,1 540,1 585,1 643 cm-1;1H NMR(400 MHz,DMSO-d6): 2.22(s,3H),7.06(d,J=7.8 Hz,2H),7.20(d,J=7.6 Hz,2H),7.40(d,J=8.0 Hz,2H),7.64~7.70(m,4H),7.91(s,1H),8.00(d,J=8.0 Hz,2H)ppm;13C NMR(100 MHz,DMSO-d6): 21.0,129.0,129.3,129.4,129.5,131.6,131.7,131.8,135.3,135.6,136.8,138.3,138.7,138.8,141.5,144.7,146.0,186.8,189.0 ppm;HRMS(ESI):m/ z calcd for C25H17Cl2O2S: 451.032 6[M+H]+;found: 451.031 8.
3b白色固体;熔点: 156~157 ℃;IR(KBr,v): 1 088,1 173,1 218,1 267,1 287,1 365,1 504,1 586,1 650 cm-1;1H NMR(400 MHz,DMSO-d6): 7.06~7.11(m,2H),7.31~7.41(m,4H),7.63~7.68(m,4H),7.92(s,1H),8.01(d,J=8.0 Hz,2H)ppm;13C NMR(100 MHz,DMSO-d6): 127.1,127.6,128.2,130.7,131.2,131.3,131.4,131.8,132.6,133.1,135.1,135.3,136.6,141.7,144.3,144.5,186.2,188.3 ppm;HRMS(ESI):m/ z calcd for C24H14BrCl2O2S: 514.927 5[M+H]+;found: 514.926 6.
3c白色固体;熔点: 149~151 ℃;IR(KBr,v): 1 095,1 175,1 267,1 281,1 396,1 505,1 540,1 583,1 646 cm-1;1H NMR(400 MHz,DMSO-d6): 7.06~7.11(m,1H),7.29(d,J=8.0 Hz,1H),7.37~7.40(m,1H),7.44(d,J=8.0 Hz,1H),7.57(d,J=8.8 Hz,4H),7.81(d,J=8.4 Hz,2H),7.91~7.97(m,3H)ppm;HRMS(ESI):m/ z calcd for C24H14Cl2NO4S: 482.002 1[M+H]+;found: 482.003 0.
3d黄色固体;熔点: 137~139 ℃;IR(KBr,v): 1 033,1 167,1 261,1 285,1 420,1 506,1 598,1 632 cm-1;1H NMR(400 MHz,DMSO-d6): 2.22(s,3H),3.78(s,3H),3.88(s,3H),6.90(d,J=8.4 Hz,2H),7.07~7.14(m,4H),7.24(d,J=7.6 Hz,2H),7.70(d,J=8.4 Hz,2H),7.85(s,1H),8.01(d,J=8.4 Hz,2H)ppm;HRMS(ESI):m/ z calcd for C27H23O4S: 443.131 7[M+H]+;found: 443.130 9.
3e浅黄色固体;熔点: 130~131 ℃;IR(KBr,v): 1 023,1 125,1 257,1 353,1 436,1 523,1 577,1 646 cm-1;1H NMR(400 MHz,DMSO-d6): 2.14(s,3H),6.94(d,J=8.0 Hz,2H),7.16(d,J=8.0 Hz,2H),7.60(d,J=8.0 Hz,1H),7.90(d,J=8.0 Hz,1H),8.01(s,1H),8.02(d,J=7.6 Hz,1H),8.22(s,1H),8.27(d,J=8.8 Hz,1H),8.40(d,J=7.6 Hz,1H),8.52(d,J=8.0 Hz,1H),8.63(s,1H)ppm;13C NMR(100 MHz,DMSO-d6): 20.7,123.8,124.0,127.1,127.3,128.8,129.3,130.1,130.7,130.8,135.1,135.3,137.3,137.3,137.6,138.0,141.5,144.6,146.9,147.1,147.9,185.8,187.8 ppm;HRMS(ESI):m/ z calcd for C25H17Cl2O2S: 451.032 6[M+H]+;found: 451.031 1.
3f白色固体;熔点: 201~202 ℃;IR(KBr,v): 1 066,1 172,1 209,1 258,1 395,1 539,1 580,1 642 cm-1;1H NMR(400 MHz,DMSO-d6): 2.22(s,3H),7.04(d,J=7.6 Hz,2H),7.20(d,J=7.2 Hz,2H),7.53~7.59(m,4H),7.81(d,J=7.6 Hz,2H),7.88~7.94(m,3H)ppm;HRMS(ESI):m/ z calcd for C25H17Br2O2S: 540.929 6[M+H]+;found: 540.928 7.
2 结果与分析
2.1 条件筛选
模板反应如图5所示,从0.5 mmol的1a和1.0 mmol的2a出发,在N,N-二甲基甲酰胺(DMF)做溶剂,50 ℃微波辐射条件下考查碱性催化剂对反应的影响.
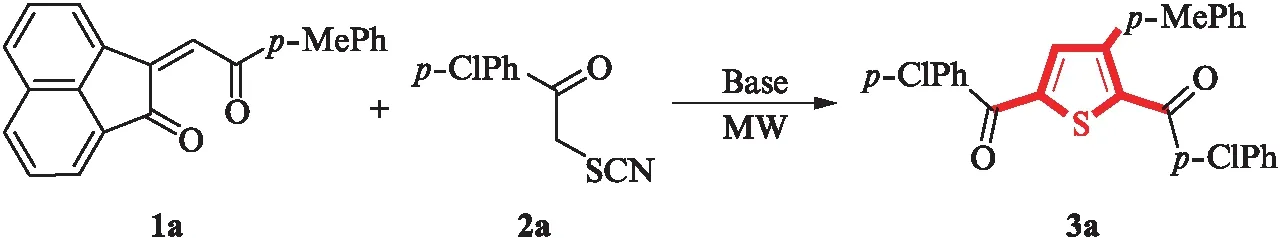
图5 模板反应Fig.5 Template reaction
结果表明,当用三乙胺(Et3N)为碱促进剂时,产率<1%.换成碱性稍强的碳酸钾(K2CO3)和碳酸铯(Cs2CO3)时,分别以13%和20%的分离产率得到3a.继续增强碱性,使用氢氧化钠(NaOH)和乙醇钠(EtONa)时,产物3a的产率也随之提高.当以叔丁醇钠(t-BuONa)为碱促进剂时,3a的产率达到了73%(表1f).选用碱性更强的叔丁醇钾(t-BuOK),产率降至64%(表1g).然后,对不同的溶剂进行筛选.乙腈(MeCN)和乙醇(EtOH)的产率均不乐观(表1h和表1i).温度由50 ℃升高到55 ℃,产率达到了83%(表1j).继续升至60 ℃,产率下降到72%(表1k).为证明微波反应比常规加热更具优势,将模板反应在常规加热条件下进行了对比,反应24 h后仅以53%的产率得到3a(表1l).综上,反应在55 ℃微波辐射和t-BuONa促进下的DMF溶液中产率最佳.

表1 条件筛选1)
2.2 底物拓展
先考察1中Ar1的多样性.如图6所示,1中Ar1无论是给电子取代基(p-MePh)还是吸电子取代基(p-BrPh、p-NO2Ph),均不影响反应的顺利进行,以75%~83%的产率合成3a~3c.
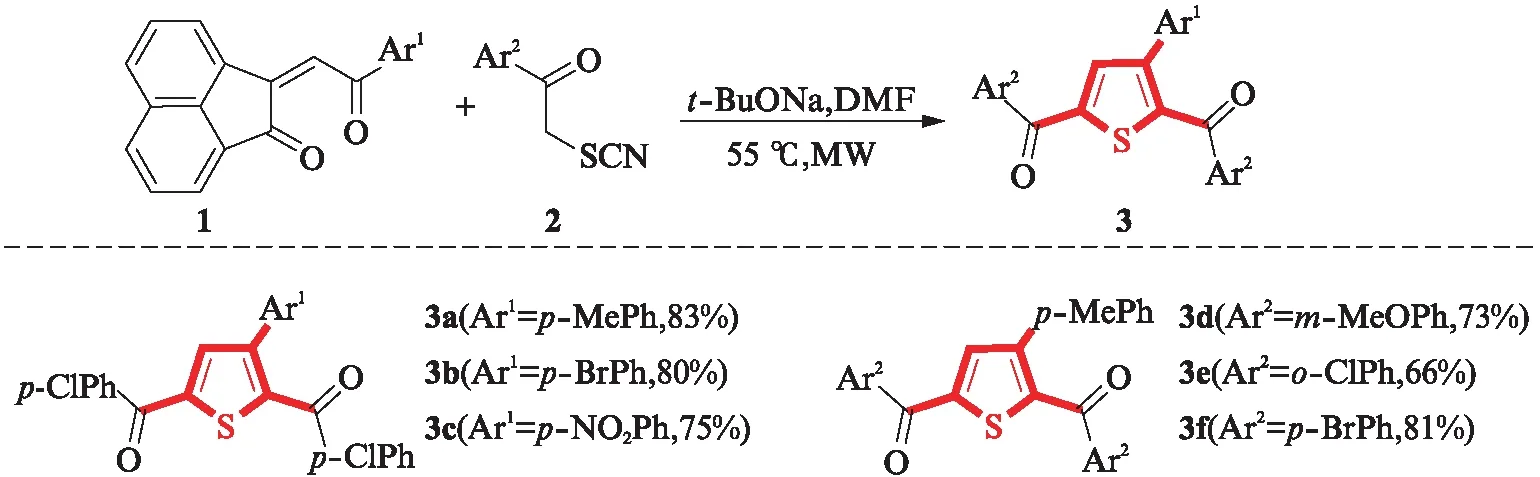
图6 2,3,5-三取代噻吩3的合成Fig.6 Synthesis of 2,3,5-trisubstituted thiophenes 3
接下来,考察2中Ar2的多样性.结果显示,原料2中Ar2取代基的位置由间位(m-MeOPh)拓展为邻位(o-ClPh)和对位(p-BrPh)时,都合成了相应的2,3,5-三取代噻吩3d~3f.
由3a与3b的产率对比可知,Ar1为给电子取代基(p-MePh)比吸电子取代基(p-BrPh)产率略高.由3a与3e的产率对比可知,Ar2为对位取代基(p-ClPh)比邻位取代基(o-ClPh)产率略高.由3b与3c的产率对比可知,Ar1为强吸电子取代基(p-NO2Ph)比弱吸电子取代基(p-BrPh)产率略低.由3a与3f的产率对比可知,Ar2为强吸电子取代基(p-ClPh)比弱吸电子取代基(p-BrPh)产率略高.
2.3 单晶测试
3a的晶胞图如图7所示,证实了产物的结构.

图7 3a的晶胞图Fig.7 Unit cell diagram of 3a
2.4 机理研究
该反应的可能机理如图8所示.芳酰基取代的苊酮1和硫氰基取代的芳酮2在碱的作用下,经连续的失质子、[3+2]环加成、原位开环、互变异构、消除、SN2反应、脱水等步骤,最终生成多取代噻吩衍生物3.

图8 可能的反应机理Fig.8 Possible reaction mechanism
3 结 语
以芳酰基取代的苊酮和硫氰基取代的芳酮为起始原料,在微波辐射和碱促进下,高效合成了6例2,3,5-三取代噻吩,选择性地修饰了噻吩骨架的C-2位、C-3位和C-5位.用时短、原料无味、产率高是该反应的显著优点,丰富了该衍生物的绿色合成方法.
猜你喜欢
煤炭学报(2022年11期)2023-01-07
陶瓷学报(2021年1期)2021-04-13
石油与天然气化工(2020年1期)2020-04-16
上海建材(2019年1期)2019-04-25
石油与天然气化工(2019年1期)2019-03-06
无机化学学报(2019年2期)2019-02-27
中国洗涤用品工业(2017年2期)2017-04-16
中国洗涤用品工业(2016年2期)2016-02-28
烟草科技(2015年8期)2015-12-20
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
