
脯氨酰羟化酶抑制剂治疗肾性贫血的研究进展
2021-05-21 13:05郑守军徐浩铜
攀枝花学院学报 2021年2期
郑守军,魏 巍,徐浩铜
(攀枝花学院 医学院,四川 攀枝花 617000)
慢性肾脏病 (chronic kidney disease, CKD) 已对全球公共卫生构成巨大威胁,在我国大约有1.2亿CKD患者,并且在成人人口中发病率高达10.8%[1-2]。贫血是CKD患者的一种常见并发症,且随肾功能下降发生率增高。据调研,我国CKD透析患者贫血患病率高达98.2%,非透析患者贫血患病率为52.1%[3-5]。目前,贫血症的治疗手段包括注射红细胞生成刺激剂 (erythropoiesis stimulating agents, ESAs)、补铁和输血[6]。这些治疗方式虽然能够改善患者的贫血,但是有些不良反应限制了它们的临床应用:ESAs能使体内半合成超生理剂量的促红细胞生成素 (EPO),这让卒中、死亡、充血性心力和心肌梗死衰竭一系列严重不良反应的发病风险增加,同时也让超敏反应、癫痫和高血压的发病风险上升[7];多数患者在服用铁剂后会出现严重的胃肠道反应甚至过敏,而且铁剂补充铁的量通常会超过人体的负荷,使人体内的铁过载和非转铁蛋白结合铁增加,从而导致氧化损伤的扩大和增加感染风险[5];输血会引发溶血反应、过敏、发烧、急性肺损伤甚至会造成疾病传播和血液污染等[5]。因此,临床上需要开发新的疗法,以减少对CKD贫血的输血依赖性,改善患者的生活质量,避免导致与EPO相关的不良反应。近几年,国内外研究者开始探索新型的红细胞生成刺激剂—低氧诱导因子脯氨酰羟化酶 (hypoxia-inducible factor-prolyl hydroxylase, HIF-PHD) 抑制剂。
1 低氧诱导因子脯氨酰羟化酶抑制剂简介
1.1 低氧诱导因子 (HIF) 结构及生物学功能
HIF是体细胞在低氧浓度下启动的一种转录因子,广泛分布于血管内膜、心脏、脑、肾脏、肝脏等部分,调控EPO、血管内皮生长因子 (VEGF) 以及糖酵解酶等基因的转录[8-9]。HIF由HIF-α和HIF-β构成[10],这两种亚基的氨基端都存在基本螺旋-环-螺旋(basic-helix-loop-helix) 和Per-ARNT-Sim (PAS)两种结构域,这对形成异二聚体并与DNA结合是必不可少的[9]。作为活性亚基的HIF-α,基因定位于人的14号染色体q21~24区,含有826个氨基酸,感受缺氧信号的活性调控区域在其两个末端,脯氨酸-丝氨酸-苏氨酸 (Pro/Ser/Thr) 的氧依赖降解结构域 (oxygen-dependent degradationdomain, ODDD) 和转录激活结构域 (transactivation domain, TAD-C)广泛存在于C末端,N末端具有TAD-N,这些结构域都是缺氧诱导蛋白转录激活、稳定和核定位的调控域,且TAD-C调整作用相对其他结构域更加精细,具有TAD-N是激活转录的必要条件。基因定位于人的1号染色体q21区的HIF-β又名芳香烃受体核转运子 (aryl hydrocarbon reeptor nuclear translocator, ARNT),是组成性亚基,不受氧浓度影响,多数情况下其表达过量[11]。
HIF-α是氧高度敏感因子,有三种亚型:HIF-1α、HIF-2α、HIF-3α,HIF-β与以上三种亚型之一组合都能诱导靶基因的不同表达[12]。HIF-1α在绝大多数组织中均有表达,对造血干细胞细胞周期调节起关键作用,是治疗缺血性疾病的潜在靶点,敲除HIF-1α基因的胚胎小鼠会因严重的心血管缺陷死亡。HIF-2α仅在特异细胞中表达,是低氧应答过程的主要介导者,参与上调EPO基因表达和低氧铁转运等。抑制HIF-1α,将导致HIF-2α量增加,从而转录功能增强,因此两者在功能上存在差异但又互相补充。DNA结合域在HIF-3α中不存在,因此不会影响基因表达,但其可以通过HIF介导的基因表达,对HIF-1α和HIF-2α的产生抑制[12-14]。
1.2 脯氨酰羟化酶 (PHD) 的结构及生物学功能
PHD属于2-酮戊二酸 (2-oxogluarate, 2-OG) (结构式见图1) 和Fe2+依赖性双加氧酶,在有氧情况下催化HIF-α特定脯氨酸残基羟基化,包括PHD1、PHD2和PHD3和PHD4四种亚型:PHD1分布在细胞核中,PHD2分布在细胞质中,PHD3在细胞核和细胞质中都有分布,PHD4主要分布在内质网中[15]。
研究表明,PHD4只在HIF-1α过表达时发挥其调节作用[16];PHD1~3因组织分布不同 (表1),功能也各不相同:PHD1~3均能羟基化HIF-α的脯氨酸残基,但其活性强度为:PHD2>>PHD3>PHD1。有研究显示,抑制PHD2可促进HIF-2α聚集,从而内源性EPO水平提高,产生的EPO是肾脏中EPO的主要来源;而PHD1和PHD3在肝脏中也对氧依赖的EPO基因转录起到一定作用[17]。PHD1基因敲除小鼠看上去正常,但其从氧化代谢转变为厌氧代谢:骨骼肌线粒体耗氧量减少,对缺血的耐受性增强,并且在运动测试中表现较差[18];PHD2基因敲除小鼠胚胎因心脏和胎盘异常导致死亡且显示血管和红细胞生成增加[19];PHD3基因敲除小鼠表现出明显的交感神经功能变化[20]。

图1 2-OG和NOG结构式

表1 PHD特性及常氧状态下的组织分布

表1(续)
1.3 脯氨酰羟化酶 (PHD)/希佩尔-林道病肿瘤抑制蛋白 (pVHL) 信号通路对HIF-α的调节
在O2、Fe2+以及2-OG存在的条件下,PHD可实现多种底物 (如HIF-α、NF-κB、激活转录因子等) 的羟基化。正常氧浓度下,PHD可以识别HIF-α的脯氨酸残基(HIF-1α: Pro402和Pro564, HIF-2α: Pro405和Pro531, HIF-3α: Pro409) 使其羟基化,经希佩尔-林道病肿瘤抑制蛋白 (pVHL) 介导,通过E3连接酶复合物泛素化途径快速降解[14];在缺氧条件下,PHD活性下降,未被羟基化的HIF-α转移到细胞核内,与HIF-β结合形成活性转录因子二聚体,再与转录共激活因子p300/Creb结合蛋白 (p300/CBP) 结合,使HIF信号通路激活,启动下游EPO转录,增加血浆中EPO水平,刺激骨髓中红细胞生成[21]。同时,低氧环境在激活HIF信号通路后,使得肝脏铁调素合成受抑制,转铁蛋白增加,从而促进铁的摄取和利用 (图2)[22-23]。
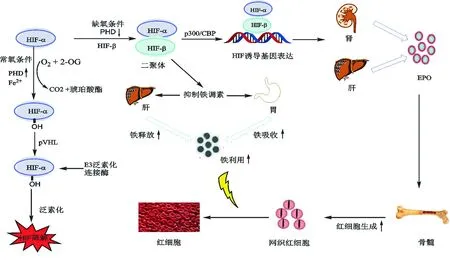
图2 PHD/pVHL信号通路对HIF-α的调节示意图
1.4 HIF-α其他调节通路
1.4.1 HIF-α上游调节通路
1.4.1.1 磷脂酰肌醇3-激酶 (PI3K) /蛋白激酶B (PKB/AKT) /哺乳动物雷帕霉素靶蛋白 (mTOR) 信号通路对HIF-α的调节
缺氧以及各种生长因子和细胞因子均可上调HIF-α蛋白表达,促进HIF-α靶基因表达,同时这些细胞因子可与相应的酪氨酸酶受体结合,激活PI3K、ATK以及mTOR信号转导通路[24]。AKT磷酸化活化后能促进叉头状转录因子家族、正性调节核因子κB和CREBP以及促凋亡分子BAD (Bcl-2 /Bcl-xL associated death promoter) 磷酸化,增强抗凋亡基因转录,增强抗凋亡因子蛋白活性、降低凋亡因子表达[25],AKT的过度磷酸化能够使细胞生长繁殖增快[26]。mTOR的C端与PI3K催化结构域同源, AKT活化后可促进mTOR的表达,高表达的mTOR可抑制AKT活性,mTOR对细胞增殖、凋亡以及成瘤具有重要调节作用,是磷酸化AKT下游重要效应分子[27]。mTOR通过5’端寡聚嘧啶核苷酸序列促使HIF-α mRNA翻译速率增快,从而促进HIF-α表达[28]。
1.4.1.2 细胞外信号调节激酶 (ERK)/丝裂原活化蛋白激酶 (MAPK) 信号通路对HIF-α的调节
ERK/MAPK信号通路在生物调节过程中起着至关重要作用,例如细胞的增殖、分化、组织侵袭及转移。MAPK路径包含MAPK激酶激酶、MAPK激酶及MAPK三个主要激酶,ERK1,2为低进化的广泛存在的丝氨酸苏氨酸激酶,其在正常和病理状态下调节细胞信号,ERK的过度表达在肿瘤的发生发展中其了至关重要的作用。Ras/Raf/MAPK(MEK)/ERK是MAPK相关通路中最为重要的信号联级,Ras激活后通过该信号联级激活下游蛋白,HIF-α是诸多ERK/MAPK信号通路调节靶点中的重要成员[29]。体外实验证明HIF-α通过p42、p44、p38α和p38γ激酶使自身磷酸化来显著增强活性,但HIF-α活性增强后并没有明显增加HIF-α蛋白的表达量[30]。ERK/MAPK信号能增强HIF-α蛋白活性,调节相应基因及蛋白的表达,并最终引起细胞增殖、凋亡、侵袭等相关生物学变化。
1.4.1.3 Bcl-2相关性抗凋亡基因3 (BAG3)/热激酶蛋白70 (HSP70)蛋白酶体途径对HIF-α的调节
BAG3隶属于BAG家族,它与HSP70结合成蛋白酶体,作用是促进线粒体细胞凋亡。BAG3蛋白的表达大多发生在胞质,胞质中的HSP70蛋白上有BAG3蛋白结合位点,BAG3与胞质中的HSP70相对应的位点结合,进而促使HSP70与Bax (Bcl-2 associated X protein) 蛋白结合,最终形成复合体(BAG3-HSP70-Bax),这样可使细胞中游离状态的Bax蛋白含量降低;由于细胞线粒体附着有Bax蛋白,Bax蛋白含量降低可导致线粒体膜电位降低和线粒体通透性的增强从而加速线粒体裂解,最终导致细胞凋亡[31]。HSP70蛋白能通过20S和26S蛋白酶体促进HIF-α蛋白降解[32],而BAG3可与HSP70形成蛋白酶体复合物,这样可减少HSP70蛋白量,从而抑制其对HIF-α的降解作用,以此促进HIF-α表达[33]。体外研究表明,非特异性蛋白酶体抑制剂MG132可使肿瘤细胞中HIF-α表达量升高[34],进一步证明了BAG3-HSP70蛋白酶体途径抑制HSP70对HIF-α的降解作用。
1.4.2 HIF下游调节通路
调控靶基因的转录是HIF-α的下游调节的主要途径,目前发现,血管内皮生长因子(vascular endothelial growth factor,VEGF)、血小板衍生生长因子(Platelet derived growth factor,PDGF)、葡萄糖转运蛋白1(glucose transporter 1)、促红细胞生成素(EPO)、内皮素1(Endothelin 1)、NO合酶2等均为HIF-α下游的靶基因,这些靶基因含有共同的启动因子HRE,HIF-α蛋白能与靶基因HER序列特异性结合,对相应靶基因的转录起到促进作用[35]。研究证明,在缺氧条件下HIF-α靶基因表达量随HIF-α表达增加而明显增加[36]。
2 小分子口服HIF-PHD抑制剂研究进展

图3 小分子HIF-PHD抑制剂
2.1 HIF-PHD抑制剂的结构特点
HIF-PHD属于2-OG加氧酶,其作用是将氧分子中的氧原子直接引入其底物中[37]。N-草酰甘氨酸 (N-oxalylglycine, NOG) (见图1) 与2-OG具有结构和极性相似,NOG类似物代替2-OG与Fe2+形成双配位结合,由于其结合力相比2-OG更强,同时NOG酰胺结构降低了邻位羧基碳原子的电子云密度降低,增加了氧亲核进攻的难度,故使得PHD不能羟基化HIF,从而达到抑制HIF-PHD的效果[12]。目前国内外已上市或在研小分子HIF-PHD抑制剂结构式见图3,由图3可见FG-2216、罗沙司他、Daprodustat、Vadadustat、Enarodustat、Desidustat都保留了NOG侧链。
2.2 小分子HIF-PHD抑制剂
HIF-PHD抑制剂通过抑制PHD来稳定HIF、刺激内源性EPO产生、上调转铁蛋白受体表达、增加铁吸收、促进红细胞成熟的小分子药物[12,14],可以有效治疗和预防HIF和EPO相关病症,如贫血、局部缺血和缺氧的病症。HIF-PHD抑制剂给CKD贫血治疗带来革命性的改变,目前仅罗沙司他已在中国上市,其余药物都在临床研究阶段。下面将对目前已上市和在研的主要HIF-PHD抑制剂的研究进展和临床疗效进行简要介绍 (表2)。
2.2.1 FG-2216
FG-2216是FibroGen公司开发的第一代HIF-PHD抑制剂,用于镰状细胞血症、肾性贫血、化疗诱导的贫血在内的各种贫血的潜在治疗。1999年5月,FG-2216作为第一种口服HIF-PHD抑制剂进入临床试验阶段[38]。在不给予ESAs治疗的情况下,FG-2216能够促进EPO基因表达上调,增加内源性EPO水平,其对PHD2的IC50为3.9 μmol/L,半衰期为15.7±1.1 h。但在Ⅱ期临床研究中出现一例暴发性肝炎致死,随即FDA全面暂停该药物的临床试验,后期证明该死亡病例为非药物引起,在2008年该药物的临床研究再次获得FDA 批准,但目前并无进一步临床研究报道。

表2 小分子HIF-PHD抑制剂治疗CKD贫血的研究进展

表2(续)
2.2.2 罗沙司他 (Roxadustat)
罗沙司他 (FG-4592) 是由FibroGen,Astellas Pharma和AstraZeneca合作开发的4-羟基异喹啉类化合物。2018年12月,NMPA以优先审评审批通过罗沙司他 (商品名:爱瑞卓) 用于治疗正在接受透析的CKD贫血患者。2019年8月,罗沙司他非透析依赖性CKD贫血治疗的新适应症获批。罗沙司他是中国率先获批上市的first in class新药,疗效不劣于现有标准疗法且具有口服的便捷性。
罗沙司他是第二代HIF-PHD抑制剂,与FG-2216相比,药动学和药效学有所改进。罗沙司他血浆半衰期约12 小时,临床研究中的给药方式是2-3次/周,每次口服1-2 mg/Kg[19],它能短暂、可逆的稳定HIF-α,使机体在不缺氧的前提下转录出相关基因,降低铁调素水平,升高内源性EPO水平,上调转铁蛋白受体表达、增加铁吸收、促进红细胞成熟,从而发挥治疗贫血的作用[39-40]。
中国开展的两项III期临床研究比较研究了罗沙司他对长期透析和未透析CKD贫血患者的疗效和安全性,305例长期透析CKD贫血终末期患者中与对照组(阿法依泊汀)相比,罗沙司他对透析患者的临床获益和副作用没有显著差别[41];罗沙司他对154例未透析的CKD贫血患者有显著的临床疗效,能有效纠正和维持CKD贫血患者Hb水平,且安全性和耐受性良好[42]。罗沙司他常见的不良反应为腹泻、头痛、背痛和疲劳;另外临床试验中有7%的出现了高血压,虽然远低于ESAs组[43]
2.2.3 Daprodustat
Daprodustat (GSK1278863) 是GSK公司研发的单环吡啶酮衍生物,已在日本和高加索地区完成临床Ⅲ期研究。2020年6月,GSK在日本批准Daprodustat用于治疗肾性贫血的新药上市申请。
Daprodustat对PHD 3种亚型的半抑制浓度IC50分别为PHD1: 3.50.6 nM; PHD2: 22.213.4 nM; PHD3: 5.5 5.1nM,其主要通过CYP2C8在肝脏中代谢,半衰期为4小时。Ⅱ期临床研究结果显示,Daprodustat组内源性EPO造成的Hb水平升高显著低于ESAs组,而且Daprodustat安全耐受性良好,血浆VEGF浓度没有显著升高;Daprodustat还可导致总胆固醇、低密度脂蛋白和高密度脂蛋白降低[7]。III期临床研究纳入271例依赖透析的患者进行了为期52周的研究,结果显示口服Daprodustat在40到52周内的平均Hb水平上达到了阿法依泊汀静脉注射液的非劣效性主要终点,在Daprodustat治疗组中发生至少一种不良事件的患者百分比为93%,在对照组中为97%。治疗组与对照组常见的不良事件是鼻咽炎 (42% vs 49%),胃肠道事件 (46% vs 46%) 和分流狭窄 (14% vs 15%)[44]。
2.2.4 Vadadustat
Vadadustat (AKB-6548) 是由Akebia Therapeutics和日本田边三菱公司联合开发的以吡啶为母核的口服HIF-PHD抑制剂。2019年7月,Akebia与三菱已向日本厚生劳动省共同提交了Vadadustat用于治疗CKD贫血症的新药上市申请,2020年8月批准上市。
Vadadustat对PHD2的IC50为0.029 μmol/L,对HIF-2α的稳定性优于HIF-1α,血清EPO升高水平随剂量依懒性增加,在口服约18 小时后血清EPO达到峰值[20],半衰期为4.5 小时[13],耐受性良好,可显著增加Hb水平,可提高网织红细胞和总铁结合的能力,降低血清铁蛋白和铁调素水平,没有观察到血压、VEGF、C-反应蛋白和总胆固醇的显著变化,最常见的不良反应为鼻咽炎、腹泻、分流管狭窄和便秘等[45-46]。
在名为J01和J03的两项研究中,评估了Vadadustat和阿法依泊汀对非透析依赖和透析依赖的CKD患者的疗效和安全性,304名非透析依赖和323名透析依赖CKD贫血的日本受试者进行了为期52周的治疗,Vadadustat治疗组Hb水平与对照组阿法依泊汀相比,都能达到预先规定的非劣性标准[47]。
2.2.5 Enarodustat
Enarodustat (JTZ-951) 是Japan Tobacco开发的一种三氮唑并吡啶类口服HIF-PHD抑制剂。Japan Tobacco在日本已完成Enarodustat针对肾性贫血的Ⅲ期临床试验,于2020年9月日本获批上市,我国药企信立泰获得该品种的中国开发授权。
Enarodustat对PHD2的IC50为0.22 μmol/L[48],在非透析性CKD贫血患者中的有效性、安全性和维持剂量研究显示,初次使用ESA (矫正组) 和稳定剂量的ESA (转化组) 患者被随机分配,以双盲方式每天一次接受2 mg、4 mg或6 mg Enarodustat或安慰剂治疗6周(1阶段),然后进行24周的开放Enarodustat治疗,以维持其Hb水平在10.0-12.0 g /dL的目标范围内 (第2阶段)。结果显示,在矫正组中,每周Hb水平的增加速率呈剂量依赖方式增加。在第1阶段中,Enarodustat组和安慰剂组在Hb水平保持在基线的±1.0 g / dL以内,无显著差异。在第2阶段的治疗结束时,两组中超过70%的受试者将Hb水平保持在目标范围内。校正组和转化组的平均处方剂量分别为3.58和3.74 mg/天。Enarodustat与铁调素和铁蛋白的减少以及总铁结合能力的增加有关,可以纠正和维持未透析的CKD贫血患者的血红蛋白水平,并且通常被很好地耐受[49]。
2.2.6 Desidustat
Desidustat是印度Zydus Cadila公司研发的一种新型口服HIF-PHD抑制剂,用于治疗接受透析和非透析的CKD贫血患者,Desidustat对PHD2的IC50为11.20 μmol/L,在肾性贫血和炎症性贫血的大鼠模型中能够持续性诱导EPO产生[50]。
健康志愿者口服给药的Ⅰ期临床研究结果显示Desidustat耐受性良好,安全有效,血浆EPO平均水平呈剂量依赖性增加,Desidustat的代谢不会因为肾脏病变的严重程度而发生变化[51]。II期临床研究共纳入了117例非透析CKD贫血患者,经过6周的治疗,与安慰剂相比,在3个Desidustat剂量水平组,主要有效性终点Hb水平的升高均实现了有统计学意义的显著差异,次要终点Hb反应率在3个Desidustat剂量水平均超过60%。安全性方面,没有观察到严重不良事件,生命体征、心电图参数或安全化验值均无明显变化[52]。
Zydus已启动了两项Desidustat III期临床试验,计划纳入未接受透析的CKD患者和正在接受透析的CKD患者。
2.2.7 Molidustat
Molidustat (BAY-85-3934) 是由Bayer公司研发的口服HIF-PHD抑制剂。Molidustat原本是Bayer的心脏病药物,目前已完成慢性肾病或终末期肾病相关贫血Ⅱ期临床试验,有望进入Ⅲ期临床。
Molidustat可以抑制3种PHD亚型 (IC50 PHD1: 0.48μM; PHD2: 0.28μM; PHD3: 0.450 μM)[45]。在肾性贫血小鼠模型中,Molidustat除纠正贫血以外,还有降血压的作用,能使血压稳定在正常水平;它还能纠正多发性关节炎大鼠模型中炎症引发的贫血,降低单核细胞趋化蛋白-1和铁调素mRNA的表达[12,53]Molidustat平均半衰期是7~13小时[54],其主要代谢产物为无药理活性的N-葡萄糖醛酸苷[55]。在三项共计纳入400多例CKD患者为期16周的随机IIb期剂量学研究中,Molidustat与安慰剂相比,可纠正Hb水平或维持在与那些继续接受ESAs治疗的患者相当的水平,副作用可控[56]。
2.2.8 其他HIF-PHD抑制剂研究进展
恒瑞医药于2019年1月28日偕同同苏州市盛迪亚生物技术和中国药科大学向国家食药监局提交DDO-3055片临床研究申请办理获审理;2018年11月,东阳光药HEC53856被默认许可临床试验,此外,三生制药的产品正在进行临床审评审批。
3 HIF-PHD抑制剂的潜在风险
HIF-PHD抑制剂最常见的不良反应是恶心、腹泻等胃肠道反应,Hb水平过分增高也是其使用的一个潜在风险[57],HIF-PHD抑制剂的致癌风险也是未来需要关注的重点,其可诱导VEGF基因的转录和表达,从而促进血管形成、提高了肿瘤细胞的供给、帮助其存活,甚至通过血管通透性增加了肿瘤转移的风险[58],此外,VEGF还与眼内新生血管疾病有关,包括年龄相关性黄斑变性、脉络膜新生血管和糖尿病视网膜病变[59]。由于HIF-PHD抑制剂能刺激红细胞的生物合成,提高个体氧的运输能力,因此很可能会被滥用于体育运动中[30]。
4 结语
HIF-PHD抑制剂是一类通过模拟机体在高海拔地区或缺氧条件下,靶向抑制PHD来稳定HIF,进而刺激内源性EPO产生且维持其正常生理水平,上调转铁蛋白受体表达、增加铁吸收、促进红细胞成熟的新型治疗贫血适应症的药物。相比注射ESAs和输血,HIF-PHD抑制剂口服的给药方式避免了前者注射或静滴给药的不便和冷藏要求,对正在接受透析和未透析的CKD贫血患者更加受益。虽然就目前研究来看,HIF-PHD抑制剂优点突出,但对其潜在的安全性问题不容忽视。未来临床研究,仍需长期评估传统ESAs和新型HIF-PHD抑制剂的治疗效果,评估HIF-PHD抑制剂对肿瘤、心血管事件、高血压和骨质疏松等潜在风险。另外,已上市和在研药物的临床研究显示,目前的HIF-PHD抑制剂缺乏对PHD亚型的特异性选择,因此,开发特异性选择的HIF-PHD抑制剂是未来研究工作的重点。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国医学物理学杂志(2022年9期)2022-10-09
中国现代医生(2022年19期)2022-08-25
现代临床医学(2022年3期)2022-06-06
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
医学概论(2022年4期)2022-04-24
家庭医学·下半月(2020年6期)2020-01-07
当代陕西(2019年23期)2020-01-06
健康大视野(2018年17期)2018-12-28
