
苦茶全长转录组测序及基因结构分析
2021-05-19 07:31庞丹丹刘玉飞孙云南田易萍宋维希陈林波
西北农业学报 2021年4期
庞丹丹,刘玉飞,孙云南,田易萍,宋维希,陈林波
(1.云南省农业科学院 茶叶研究所,云南勐海 666201;2.云南省茶学重点实验室,云南勐海 666201)
苦茶(Camelliasinensis)是中国的一种特异茶树种质资源[1],由其制成的茶叶感官品质表现极苦,且与其他茶类的苦味不同,因此,被命名为苦茶。苦茶多分布于云南、四川,以及广东、湖南、江西毗邻区,尤以云南以及南岭山脉两侧最多[2-3]。在苦茶的原生地,苦茶成为当地人民生活的必须品,将它作为一种药物饮用,认为长期饮用苦茶具有“退火发汗、解毒、治病”的功效[2]。研究已证实,苦茶嘌呤生物碱的组分与常规茶树差异较大,其以苦茶碱(1,3,7,9-四甲基尿酸)为主,其次是咖啡碱、可可碱[3-4],苦茶碱具有镇静催眠[5]、抗抑郁[6]等药理活性,并且毒理试验鉴定苦茶碱为无毒[6],其还可以削弱咖啡碱的兴奋作用[7],这使得苦茶的价值不断提升,消费需求连年增加。苦茶除含有高含量的苦茶碱外,还有非儿茶素类茶多酚[8]。同时,苦茶中还具有高含量的表没食子儿茶素(EGC),EGC含量与红茶中关键品质成分茶黄素的含量呈极显著正相关[2,9],所以苦茶可以作为选育和培育特异高级红茶茶树品种的育种材料;以往的研究表明,苦茶茶叶中存在一种具有丁香香气的特殊物质丁子香酚甙[2]。这说明苦茶还具有多种不同于常规栽培茶树的特异性状,这些性状形成的机理、性状相关基因标记的开发以及遗传机制的研究,都需要借助于苦茶资源。
当前,大多数转录组测序是基于Illumina平台的第二代测序(SGS)技术生成的[10]。但是,SGS技术不能产生较长的转录本,并且SGS无法获得可变剪接等信息,从而限制了该技术在转录组测序中的利用[11]。而全长转录组测序是基于第三代测序(TGS)平台进行的,该方法可以产生长读段,因此可以直接测序全长转录本[12],在这方面全长转录测序优于短读测序;另外,它还可用于选择性剪接事件及初级-前体-成熟RNA结构的分析,以帮助更好地理解RNA的加工过程;此外,其可以在转录水平上全面分析由交替剪接和基因融合产生的结构变异,比SGS更准确地检测结构变异(SV),使其适合于多倍体物种或具有高重复序列和高杂合性的物种的转录组分析[13]。由于茶树基因组具有高的重复序列(至少包含64%的重复序列)和高的杂合度[14],以及苦茶特异性状形成机制需进一步解析。因此,本研究以苦茶为材料,利用PacBio平台进行Iso-Seq,对所获得序列与参考基因组比对、同时对其进行功能注释、基因结构分析等,为苦茶特征形状遗传分析提供基础数据,为更好地了解和利用这一重要的特异茶树资源提供帮助。
1 材料与方法
1.1 试验材料
用于测序分析的苦茶资源‘老曼娥苦茶’来源于云南省农业科学院茶叶研究所试验基地,其6个不同组织(芽、叶、花芽、花蕾、茎和幼果)的样品采摘后用液氮冷冻,并置于-80 ℃冰箱,之后送至诺禾致源科技股份有限公司(北京)进行测序。
1.2 RNA提取与构建文库
参照张亚真等[15]的方法,分别提取苦茶芽、叶、花芽、花蕾、茎、幼果不同部位的总RNA,检测合格后进行等量混匀。利用带有Oligo(dT)的磁珠从混匀后的RNA样品中分离出mRNA,并利用SMARTer PCR cDNA Synthesis Kit将其反转录为cDNA,接着用BluePippin对cDNA进行片段筛选,对全长cDNA进行损伤修复、末端修复,并连接SMRT哑铃型接头,最终使用核酸外切酶消化获得文库。
1.3 Iso-Seq及组装分析
采用软件SMRT Link v5.0对输出进行过滤和处理,参数:--minLength=200,--minReadScore=0.75,最终得到的数据即为有效数据,校正后获得CCS序列(Circular Consensus Sequence,参数:--minPasses=1,minPredictedAccuracy=0.8);通过检测CCS是否包含5′-primer、3′-primer、poly-A对CCS进行分类找出FLNC(full-length non chimera)序列,之后对FLNC进行聚类和校正,最终获得polished consensus序列用于后续分析。使用MISA(http://pgrc.ipk-gatersleben.de/misa/misa.html)软件对单、二、三、四、五和六核苷酸的最低重复次数分别设置为10、6、5、5、5和5,进行SSR搜索。
1.4 参考基因组比对及功能注释
使用GMAP(http://research-pub.gene.com/gmap/)将三代测序reads比对到物种的参考基因组[16],进行饱和度曲线及转录本密度分析,并对未比对到参考基因组上转录本进行七大数据库(NR、NT、Pfam、KOG/COG、Swiss-Prot、KEGG、GO)的功能注释。
1.5 基因结构分析
根据转录本与参考基因组比对结果,对转录本进行再次校正,然后进行利用SUPPA(https://bitbucket.org/regulatorygenomicsupf/suppa)、Tapis(https://bitbucket.org/comp_bio/tapis)等软件进行可变剪切分析、APA预测、新基因和新转录本鉴定、新基因数据库注释、转录因子分析。并利用CNCI(https://github.com/www-bioinfo-org/CNCI)、PLEK(https://sourceforge.net/projects/plek/)、CPC(http://cpc.cbi.pku.edu.cn/)软件以及Pfam数据库对PacBio测序数据进行编码潜能预测,并将最终得到的LncRNA进行后续分析。
2 结果与分析
2.1 Iso-Seq分析
2.1.1 测序结果与组装全长 转录组测序共获得8 730 808个Subreads(10G),平均Subreads的长度为1 145 bp,N50为1 741。通过校正,CCS序列(环形一致性序列)有418 424个,含有5′端引物的reads数有338 306个,含有3′端引物的reads数目为368 458个,含有PolyA尾的reads数目为351 952个;全长序列数目为 295 803个,FLNC序列的数目有217 701个,且FLNC的平均长度为1 836 bp;最终获得Polished consensus序列132 334个,用于后续分析。
2.1.2 SSR分析 利用MISA对苦茶全长转录组测序获得的25 735条(大于500 bp)转录本序列进行搜索,共在12 926条转录本序列中发现符合标准的21 106个SSR位点,其中5 227个转录本含有大于1个SSR位点,SSR的发生频率为50.22%,SSR出现率为82%,平均2.4 kb出现1个SSR。在检测到的苦茶转录组SSR中,单、二和三核苷酸重复类型的比例较大,分别占总SSR的32.49%、47.91%和16.18%;其他3种重复类型所占比例相对较少,只占总SSR的3.42%,结果见表1。苦茶转录组SSR重复单元的重复次数分布在5~46,其中5~10次重复的SSR位点有13 625个,占位点总数的64.56%;11~20次重复的SSR位点有7 299个,占总数的34.58%;20次重复以上的SSR位点有0.86%(表1)。
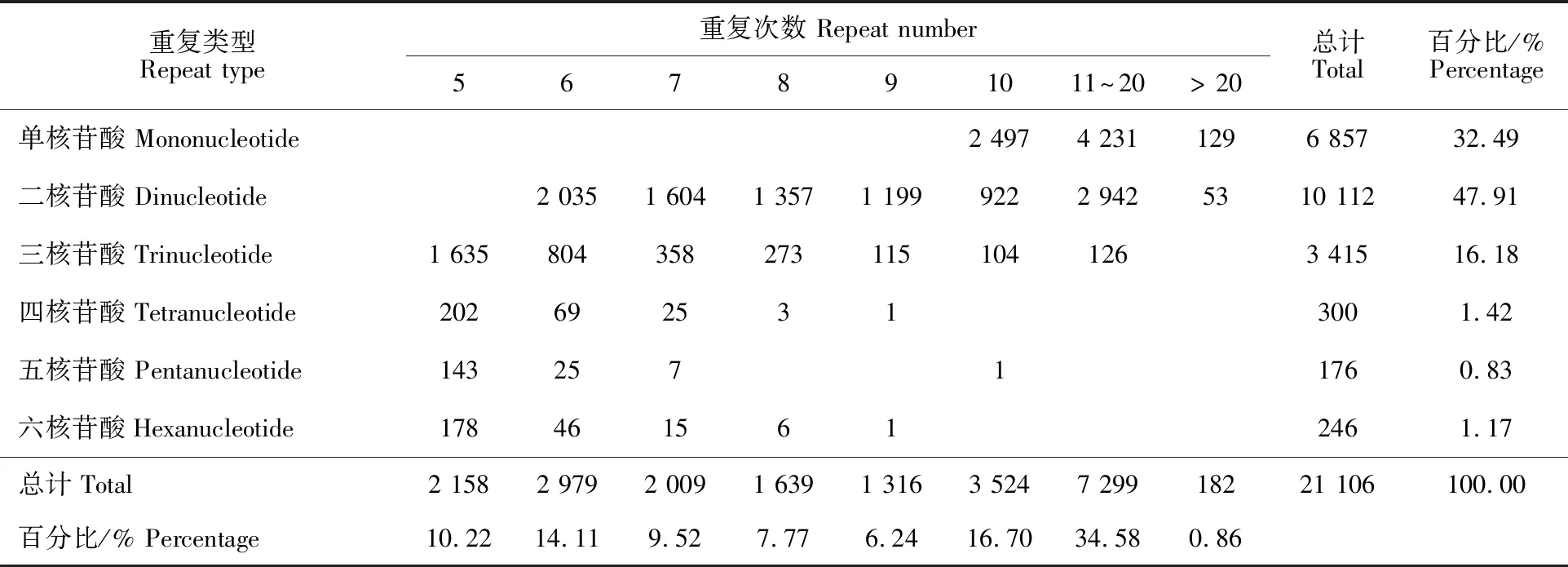
表1 苦茶SSR的类型、数量及分布频率Table 1 Type, number and frequency of SSRs in Kucha
检测到的苦茶全长转录组SSR核苷酸基序类型中(表2),二核苷酸重复基序中AG/CT (7 720个),AT/AT(1 653个)的出现频率较高;三核苷酸中出现最多的重复基序是AAG/CTT(926个),其次是ACC/GGT(474个)、ATC/ATG(446个)。四核苷酸中以AAAT/ATTT(126个)、AAAG/CTTT(47个)、AAAC/GTTT(41个)占优势;五核苷酸中分别以AAAAC/GTTTT(35个)、AAAAG/CTTTT(34个)和AAACC/GGTTT(27个)出现频率较高;AAAAAC/GTTTTT(16个)是六核苷酸中出现频率最高的,占其总数的12.5%,其次是AAAAAG/CTTTTT(14个)和AACCCT/AGGGTT(13个)。上述分析结果为下一步苦茶种质资源鉴定、遗传多样性的分析和遗传图谱的构建等研究工作提供参考。
2.2 参考基因组比对与Unmapped转录本的功能注释
分析与茶树的参考基因组的比对结果,发现能比对到基因组上的一致性序列的reads共 105 898个,依据比对情况将序列分为五种类型:Unmapped、Multiple mapped、Uniquely mapped、Reads map to ‘+’、Reads map to ‘-’,同参考基因组比对结果见图1。其中不能比对到基因组上(Unmapped)的reads有26 436个,占 19.98%;在参考序列上有多个比对位置(Multiple mapped)的reads有1 359个,占1.03%;在参考序列上有唯一比对位置(Uniquely mapped)的reads有104 539个,其中比对到基因组上正链(Reads map to ‘+’)的reads有101 781个,占76.91%;比对到基因组上负链(Reads map to ‘-’)的reads有2 758个,占2.08%。
为了获得转录本全面的功能信息,将未比对到参考基因组上的转录本进行七大数据库(NR,NT,Pfam,KOG/COG,Swiss-Prot,KEGG,GO)的功能注释,结果有11 152个转录本在NR数据库中比对到311个物种上,比对上较多的5个物种包括茶树Camelliasinensis(1 519个)、葡萄Vitisvinifera(1 413个)、胡桃Juglansregia(441个)、中粒咖啡Coffeacanephora(376个)、水芙蓉Nelumbonucifera(354个)。从NR数据库的注释结果来看,注释到茶树的基因最多,其次是葡萄;对基因进行KO注释的结果显示,苦茶转录组中有10 976个得到注释,与茶叶品质相关的KEGG通路主要有氨基酸代谢途径(409个)、苯丙烷物质的生物合成(70个),与茶叶香气有关的KEGG通路主要有萜类物质骨架生物合成(38个)、倍半萜生物合成(114个)、黄酮及黄酮醇的生物合成(2个),涉及苦茶中苦茶碱合成相关的嘌呤代谢(74个),与植物激素信号转导相关的有64个,MAPK信号通路相关的则有31个等。同时在KOG数据库中6 296个转录本得到注释,其中注释到次生代谢物合成、运输和代谢有425个,氨基酸运输和代谢的有357个,另外,还有6 221个转录本注释到GO数据库,由于同一个转录本对应到多个GO条目下,使得在GO数据库中得到注释的基因数要比注释到的转录本数目多。

表2 苦茶全长转录组中SSR基序的分布Table 2 Distribution of SSR motif in full-length transcriptome of Kucha
2.3 基因结构分析
结构分析是三代全长转录组中的一个重点内容,不同的样本转录物不尽相同,进行转录本结构分析,可以统计所有转录本的结构差异,能够准确识别二代测序中无法区分的同源基因或者同源异构体等。
2.3.1 可变剪切分析 利用SUPPA软件对数据进行分析,结果(图2)共获得4 892个基因发生了可变剪切事件,其中外显子跳跃事件(Skipped exon,SE)的基因有640个,占比为5.21%;外显子互斥事件(Mutually exclusive exon,MX)的基因有43个,占比为0.35%;内含子滞留事件(Retained intron,RI)的基因有1 635个,占比为 13.32%;5′UTR区可变事件(Alternative 5′ splice site,A5)的基因有947个,占比为7.71%;3′UTR区可变事件(Alternative 3′ splice site,A3)的基因有1 355个,占比11.04%;起始外显子可变事件(Alternative first exon,AF)的基因数目有202个,占比1.65%;终止外显子可变事件(Alternative last exon,AL)的基因数目有70个,占比 0.57%,其中内含子滞留事件占最大比例,其次是3′UTR区可变事件。
2.3.2 新基因和新转录本分析 从与茶树参考基因组比对结果来看,全长转录本可分为三类,其中比对到已知基因的已知转录本有10 842个,占比24.56%;已知基因的新转录本有26 184个,占比59.32%;比对到参考基因组未注释区域的新基因有7 115个,占比为16.12%(图3)。为了探究新基因的功能信息,同样将新基因的转录本进行七大数据库(NR、NT、Pfam、KOG/COG、Swiss-Prot、KEGG、GO)的功能注释。NR数据库比对结果显示,3 627个转录本被比对到142个物种上,比对上的数目较大的物种分别为葡萄Vitisvinifera(866个)、胡桃Juglansregia(176个)、可可树Theobromacacao(173个)、中粒咖啡Coffeacanephora(136个)、芝麻Sesamumindicum(135个)、茶树Camelliasinensis(121个)、水芙蓉Nelumbonucifera(120个)。以上的分析结果显示,注释到葡萄的基因数量最多,其原因有可能是茶树与葡萄的亲缘关系较近。
与GO数据库进行比对,结果发现有575个转录本得到了注释,在生物学过程类别中,代谢过程(259个)、细胞过程(243个)、单生物过程(176个)最多;细胞组分类别中,细胞部分(120个)、细胞(120个)、细胞器(96个)较多;分子功能类别中,结合活性(382个)、催化活性(237个)较多。根据它们参与的KEGG进行分类,结果发现新基因中有5 626个成功得到注释,苯丙烷生物合成相关的有18个、其中与苦茶中苦茶碱合成相关的嘌呤代谢有17个,与茶叶品质相关的氨基酸代谢有69个,与制成的茶叶相关的萜类化合物代谢有31个,信号转导相关的途径有150个,其中植物激素信号转导有43个。在KOG数据库中1 896个转录本被注释,按照功能一共分成24类,其中一般功能预测最多,其次是翻译后修饰、蛋白折叠和分子伴侣,之后为信号转导机制。
2.3.3 转录因子分析 本研究预测到的转录因子(TF)有1 918个,隶属于84个TF家族,将注释到转录本数目最多的前30个TF家族进行柱形图展示,分析结果如图4。在获得的TF家族中,bHLH家族有96个、C3H家族有80个、bZIP家族有70个、MYB-related家族有66个、C2H2家族有66个、MYB家族有57个。
2.3.4 LncRNA分析 经过筛选后,依据在基因组的位置对最终得到LncRNA进行分类,并对占比情况进行展示,其中基因间区(LincRNA)最多,含有569个,占比73.14;完全位于蛋白编码基因的intron区(Sense-intronic LncRNA)次之,有141个,占比为18.12%;与蛋白编码基因的exon区有overlapping(Sense-overlapping)有42个,占比5.4%;反义链(Anti-sense_LncRNA)有26个,占比3.34%(图 5-A);各软件预测为noncoding的转录本条数画成维恩图,结果展示(图5-B)。
3 讨论与结论
目前,PacBio-Iso-Seq测序手段已在多种植物中得到了广泛的应用,该方法相比于二代测序具有读长长、无需组装转录组就可以直接获得全长转录本、更准确地检测结构变异和适合于具有高重复序列和高杂合性的物种的转录组分析等优势[11-13]。本试验利用Iso-Seq技术,结合生物信息学分析,获得了苦茶全长转录组数据。
本研究共得到8 730 808个Subreads,平均Subreads的长度为1 145 bp,N50为1 741,可以看出全长转录组测序读长长且连续性较高。全长非嵌合(FLNC)reads有217 701个,通过对FLNC-reads的聚类及校正分析,最终获得polished consensus序列132 334个。庞丹丹等[17]对不同叶色的6份茶树材料进行二代测序,共获得112 233个Unigene,平均长度为759 bp,N50为 1 081 bp。Li等[18]采用RNA-seq对龙井43的根、茎、4个不同嫩度的叶片及花蕾和果实等13个组织进行测序分析,共获得347 827条Unigene(>200 bp),平均长度为791.2 bp,N50为1 342 bp。上述结果表明在获得的序列质量和基因数等方面,三代测序结果均优于二代测序。
苦茶的全长转录组测序在很大程度上补充了现有茶树基因资源,并为发现新的或以前未被识别的蛋白质的编码基因和转录亚型提供了优势。与茶树参考基因组比对,分析发现大多数PacBio转录本是已知基因的新亚型(59.32%),有 16.12%的转录本为7 115个新基因提供了证据(图3)。分析表明,新的转录组数据对探究苦茶的注释具有巨大的潜力。Unmapped转录本在NR数据库中的比对结果显示,共比对上311个物种,比对到最多的物种仍是茶树,其次是葡萄,这表明比对所用到的参考基因组可能还需进一步完善。比较KEGG(10 976+5 626)、KOG (6 296+1 896)、GO(6 221+575)3大数据库功能统计情况,发现注释到KEGG的转录本数最多(括号内数值为未比对到基因组的转录本和新转录本数量,下同),这些转录本中包含多个转录本与茶叶品质和苦茶特征性状形成相关的代谢途径,如苦茶碱等嘌呤生物碱代谢(74+17)、氨基酸代谢(409+69)、苯丙烷生物合成(70+18)和萜类物质的生物合成(152+31)等。Wang等[19]对3个苦茶品种进行转录组测序,通过与嘌呤生物碱代谢的KEGG通路富集分析,筛选出一批与苦茶碱和咖啡碱生物合成相关的关键基因。同样,本研究新注释到的一些转录本亦能够为后期对苦茶性状形成的研究提供基因资源。
可变剪切(Alternative splicing,AS),即某些基因的一个mRNA前体以不同的剪接方式,形成不同的mRNA异构体[20];其能够调控基因的表达水平和促进蛋白质组的多样性,在植物生长、发育和胁迫响应中发挥着关键作用[21-23]。选择性剪接的亚型具有基于组织或时间的优先表达特征,并且它们也受环境条件的影响[24]。在茶树中,研究发现可变剪切亚型在高温、干旱、低温等不同环境胁迫条件下的表达模式存在明显差异[25-26],同时可变剪切事件还参与了黄酮、类黄酮和花青素等重要物质的次生代谢过程中[24, 27-28]。因此,对基因结构进行分析,发现4 892个基因发生了选择性剪接,其中内含子滞留事件占比最大,这为进一步研究可变剪切在苦茶特异成分代谢中的作用奠定了基础。
以往的研究表明,转录因子(TF)参与植物多种生长代谢过程中。在茶树中,发现它们不仅参与叶片花等器官的发育[29-30],以及胁迫反应[31],还参与儿茶素[17]、茶氨酸[32]和花青素[33-34]等多种次生代谢过程。本研究还预测得到1 918个TF,在获得的TF家族中,bHLH家族有96个、C3H家族有80个、bZIP家族有70个、MYB-related家族有66个、C2H2家族有66个、MYB家族有57个。这些转录因子的预测与分析,为之后研究苦茶特异性状(苦茶碱[3-4]、丁子香酚甙[2]和高EGC[9]等)相关基因表达以及详细的基因家族分析提供数据。此外,本研究还预测得到778个LncRNA,它们可能在茶树次生代谢的调控中起着特殊的作用。
本研究为苦茶品质和特异性状形成机制的研究提供了基础数据,上述结果将有助于进一步开展苦茶特异性状相关基因标记的开发、并为其形成的机理及遗传机制的研究奠定基础。
猜你喜欢
当代化工研究(2022年17期)2022-10-12
军事文摘(2022年16期)2022-08-24
中国典型病例大全(2022年10期)2022-05-10
茶道(2022年3期)2022-04-27
云南画报(2021年10期)2021-11-24
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
家庭医学·下半月(2016年5期)2016-05-14
延河·绿色文学(2016年8期)2016-05-14
